




Oculosympathetic paresis, historically known as Horner sydrome, classically results in a triad of ptosis, miosis, and anhydrosis on the affected side. Any interruption or insult to any part of the sympathetic pathway to the eye can result in these classic findings. There are many possible etiologies along the three-neuron pathway, however, a few potentially life threatening causes must always be ruled out, especially in acute presentations. One life threatening etiology, in particular, to rule out is of internal carotid artery dissection (ICAD).
A 38-year-old white female was referred to our clinic for a second opinion by her primary care physician for a sudden onset headache, ptosis, and miosis of the left side following a short endurance race 24–48h prior. Entering visual acuities were 20/20 in the right eye (OD) and 20/20 in the left eye (OS). Emergent magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) revealed a severe left ICAD. The patient was started on oral anticoagulants and oral steroids. Spontaneous resolution occurred three months later upon confirmation with repeat MRI/MRA. This case report reviews the clinical findings, diagnoses, treatment, and management of patients with Horner syndromes secondary to ICAD.
La paresia oculosimpática, históricamente llamada síndrome de Horner, deriva clásicamente en una tríada de ptosis, miosis y anhidrosis en el lado afectado. Cualquier interrupción o lesión de cualquier parte de la vía simpática hacia el ojo puede derivar en estos hallazgos clásicos. Sin embargo, existen muchas etiologías posibles a lo largo de la vía de las tres neuronas, debiendo descartarse siempre diversas causas potenciales de riesgo, especialmente en presentaciones agudas. En particular, una etiología de riesgo a descartar es la disección interna de la arteria carótida.
Una mujer blanca de 38 años fue remitida a nuestra clínica para una segunda opinión, por parte de su médico de atención primaria, debido a la aparición repentina de dolor de cabeza, ptosis y miosis en el lado izquierdo, tras una breve carrera de resistencia durante las 24–48 horas anteriores. Las agudezas visuales a su ingreso eran 20/20 en el ojo derecho (OD) y 20/20 en el ojo izquierdo (OS). Las imágenes emergentes de resonancia magnética (IRM) y angiografía de resonancia magnética (ARM) revelaron una disección severa de la carótida interna izquierda. Se suministraron a la paciente anticoagulantes y esteroides orales. La resolución espontánea se produjo a los tres meses, tras la confirmación con nuevas pruebas de IRM/ARM. Este informe de un caso supone una revisión de los hallazgos, diagnóstico, tratamiento y gestión de los pacientes con Síndromes de Horner secundarios a las disecciones de arteria carótida interna.
Horner syndrome, commonly also referred to as oculosympathetic paresis, was first described by three American army physicians in 1864 in a soldier who was shot through the throat.1 In 1869, Johann Friedrich Horner, a Swiss ophthalmologist, described the classic triad of findings in this condition which then became known as “Horner syndrome”.1 Horner syndrome is classically described as an interruption to the sympathetic nervous system responsible for innervation to the ophthalmic region of the eyes, head, and neck.2 The normal sympathetic nervous system is responsible for ipsilateral mydriasis, normal ipsilateral lid posture/position, and ipsilateral sweating of the forehead, which includes such structures as the iris dilator muscles, Müller's muscle of the upper eyelids, and the sweatglands of the ipsilateral forehead region.2–4 As a result, a Horner syndrome classically presents as ipsilateral miosis, subtle ptosis, and anhydrosis when the sympathetic innervation to these areas is interrupted. The miotic pupil in Horner syndrome occurs secondary to the unopposed innervation of the parasympathetic system, while the ptosis and anhidrosis findings are induced by the lack of sympathetic innervation in general. With sympathetic disruption, the iris constrictor muscle is unopposed by the dysfunctional iris dilator muscle leading to anisocoria of approximately 1–2mm of miosis on the affected side, which is more pronounced in darker/scotopic conditions and less evident in brighter/photopic conditions.5 Sympathetic disruption to Müller's muscle will result in upper lid ptosis as it is directly responsible for approximately 2mm of upper lid levation.5 Sympathetic disruption to the sweatglands of the upper forehead results in lack of sweating on the affected side. Etiologies are vast and range from benign to life-threatening causes such as malignancies or internal carotid artery dissections.3,5 Some of the more common causes of Horner syndromes are outlined in Table 1.
Most common causes of Horner syndrome.1
| First order | Second order | Third order |
| - Neoplasm | - Neoplasm | - Neoplasm |
| - Hemmorrhage | - Enlarged thyroid | - ICA dissection |
| - Infarction | - Neck/thoracic surgery | - Skull base lesions |
| - Brainstem vascular malformation | - Subclavian artery aneurysm | - Cluster/migraine headaches |
| - Any pathology involving hypothalamus, brainstem, and spinal cord | - Compression of cervical sympathetic chain | - Arteritis of ICA |
| - Multiple sclerosis | - Tumors | - Aneurysm of ICA |
| - Disseminated sclerosis | - Pancoast tumor | - Cavernous sinus lesions |
| - Trauma | - Trauma | - Fibromuscular dysplasia |
| - Trauma |
Any disruption/lesion along the oculosympathetic neuronal pathway has the potential to cause a Horner syndrome. In cases that are acute/acquired, a timely investigation of the entire oculosympathetic system is imperative to rule out any underlying life-threatening lesions/complications. Internal carotid artery dissection (ICAD) is an important, life-threatening cause that must be considered in all acute/acquired Horner syndromes, especially those that are painful in nature.2
Case reportA 38-year-old white female presented to our clinic after being referred by her primary care physician (PCP) that same day for an on-going headache and a feeling of “pain/pressure” behind her left eye (OS) and ipsilateral neck region. The patient reported that her PCP had obtained a computerized tomography (CT) that morning but that it was read as “normal with no structural abnormalities.” Upon presentation, she reported that along with her headache and pressure feelings, her pupil OS appeared to be smaller than her pupil OD and that her upper lid OS was more droopy than her upper lid OD. The severe headache, pupil abnormalities, and lid droopiness started within the previous 24–48h according to the patient. She denied any recent trauma. Interestingly though, she said she had run a 5km race 24–48h prior to any of her current signs/symptoms, but that after the race she “didn’t feel right”. She reported she did have to stop mid-race to bend over and tie her shoes. Her medical history was positive only for seasonal allergies and a history of a concussion 3 years prior after falling off of her bike. The patient reported no significant allergy contributory to her presentation. As a precaution, the PCP had also started the patient on Zithromax® (Azithromycin, Pfizer, New York, NY) antibiotic capsules for any possible underlying sinus infection. The patient's family ocular history was entirely unremarkable. She denied any tobacco use and reported only drinking alcohol occasionally/socially.
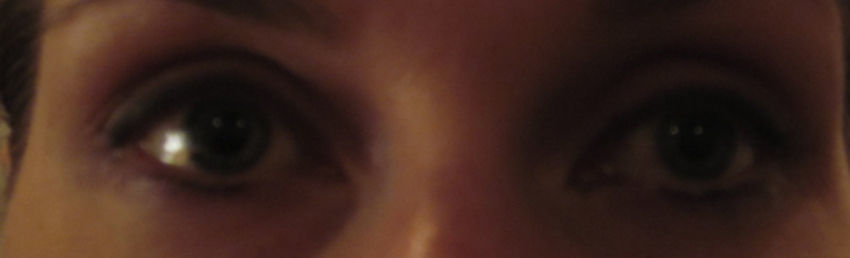
At presentation, Snellen visual acuities were 20/20 OD and 20/20 OS without correction. Her extraocular motilities were noted to have full range of motion in both eyes (OU) with no signs of restriction nor any complaints of diplopia. Confrontational visual fields were full-to-finger-count OU. Pupils were measured in light and dark conditions. In light, the pupils measured 4mm OD and 3mm OS, respectively. In dark, the anisocoria increased with the pupils measuring 6mm OD and 3mm OS. Both pupils reacted briskly to light and there were no signs of any afferent pupillary defect OU. There was no sign of light and near dissociation in either eye. Her accommodative pupillary responses were normal OU as well. Adnexal examination did reveal subtle upper lid ptosis OS when compared to the OD. Interpalpebral fissure widths were measured at 11mm OD and 8mm OS confirming the OS lid ptosis. Fig. 1 shows the presenting subtle ptosis OS with mild anisocoria. No signs of ipsilateral anhydrosis were found nor reported by the patient. The slit lamp biomicroscopy exam was entirely unremarkable OU. Intraocular pressures measured via Goldman tonometry were 14mmHg OD, and 14mmHg OS. The patient's dilated fundus exam revealed that her posterior poles and peripheral retinas were both within normal limits. Cup-to-disc ratio was 0.2 H/V OU with no signs of optic nerve edema. Common differential diagnoses to consider are listed in Table 2.
Common differential diagnoses of pupil and ptosis abnormalities (not an exhaustive list and in no particular order).6
| Condition | Clinical presenting features |
| Myasthenia gravis | Ptosis, fluctuating (waxing and waning) Sx, typically worse at end of day, worsens with exhaustion, pupils never involved, cranial nerves palsies possible, more common in younger women, associated with thalamus issues |
| Adie's tonic pupil | Ptosis never present, ocular motilities normal, pupil dilated secondary to lesion in ciliary ganglion disrupting parasympthetic innervation, anisocoria worse in brighter conditions, anisocoria usually improves with 1/8% pilocarpine |
| Myogenic ptosis | Typically bilateral, chronic, symmetric, pupils not involved, ocular motilities normal |
| Oculomotor (CN III) palsy | Ptosis, ocular motility disruption, pupil involved with aneurysmal compression, older individuals typically, history of vasculopathic causes (diabetes, hypertension, hypercholesterolemia, etc.) |
| Horner syndrome | Ptosis, miosis, anhidrosis, anisocoria worse in dark conditions, multitude of underlying causes, ocular motilities normal, acute or chronic onset (acute more concerning), younger individuals normally, heterochromia if congenital |
| Pharmacologic anisocoria | History of miotic or mydriatic agent, ocular motilities normal, acute onset, no change in anisocoria with 1% pilocarpine |
| Physiologic anisocoria | Absent ptosis, same degree/percentage of anisocoria in both dark and bright conditions, ocular motilities normal, normal pupillary reaction to light |
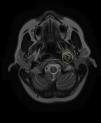
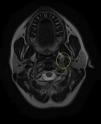
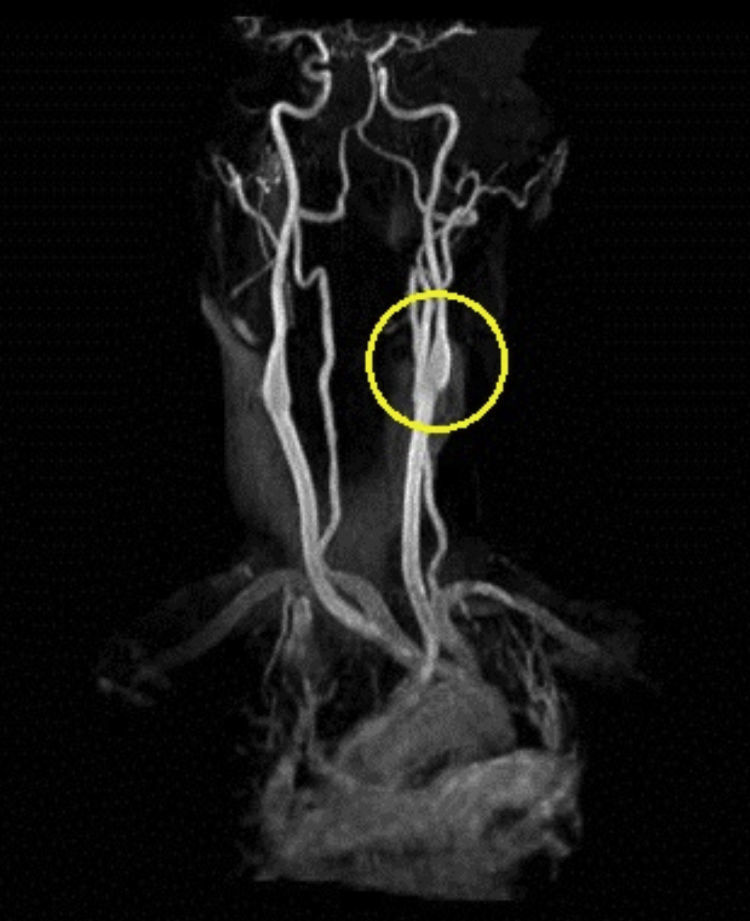
Based on the clinical presentation, the patient was diagnosed with a painful left-sided, partial Horner syndrome of unknown etiology based on the anisocoria and ptosis OS findings, however carotid artery dissection was highly suspected given the painful nature of presentation. Emergent imaging studies (including MRI/MRA, chest X-ray, and CT scans) were arranged through the PCP that same day. Four days later the PCP notified our clinic that the imaging studies had, indeed, revealed a carotid artery dissection of the distal half of the left internal carotid artery (ICA) at the level of approximately cervical vertebrae C3. Figs. 2–4 highlight the dissection of1 the left internal carotid artery via standard MRI and MRA which was classified as “severe” by the radiologist and scored as an “8 or 9 on a scale to 10”. The PCP had urgently started the patient on 325mg of aspirin daily and referred the patient to a neurologist for further consultation.
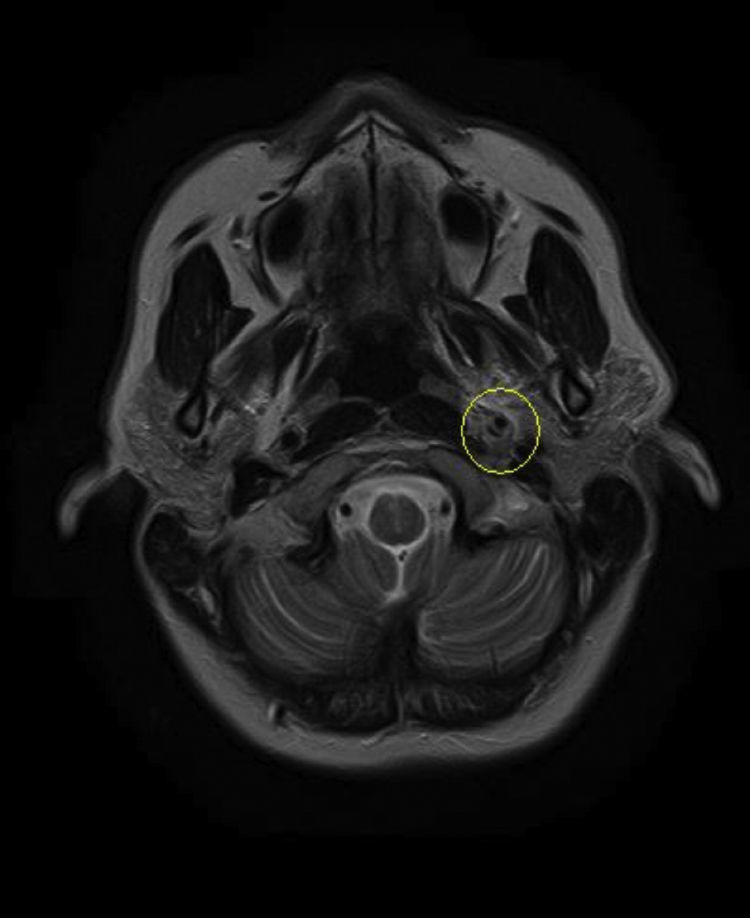
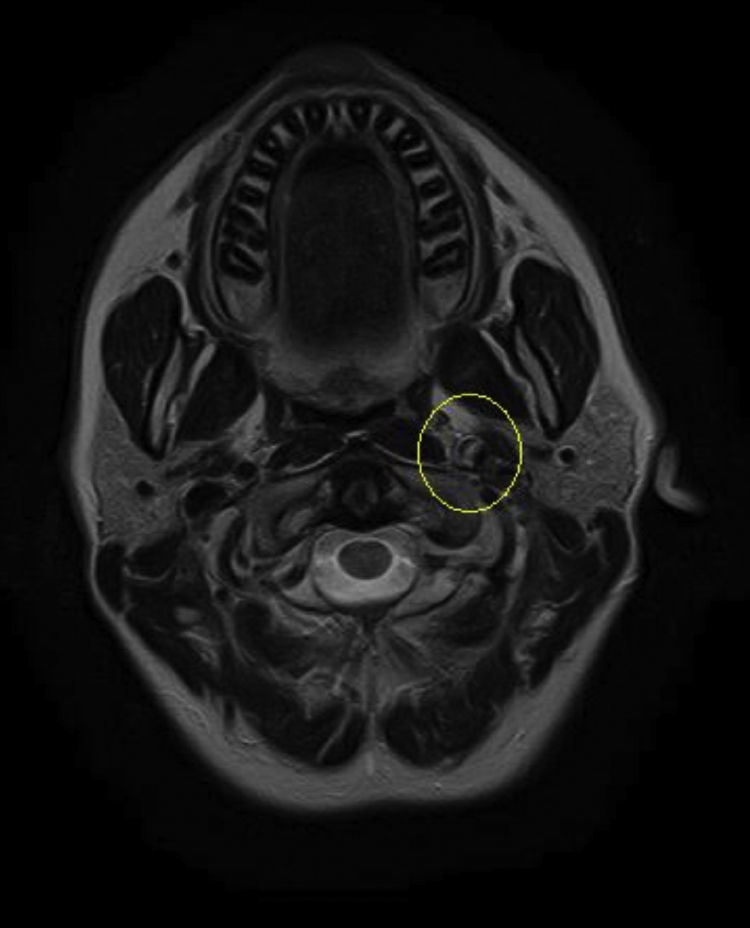
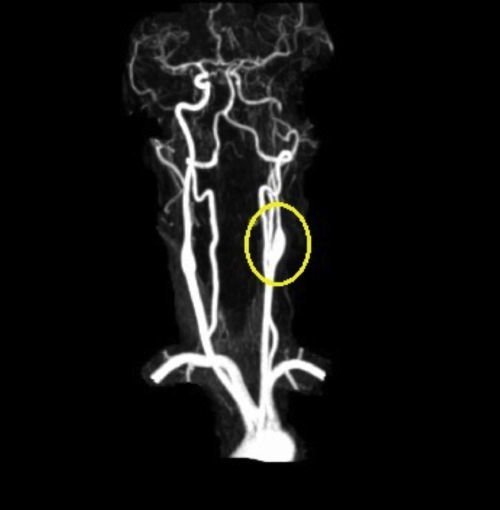
One week later the patient returned for follow-up. She reported she was feeling better but that the headache was still present, although not as severe as the previous week. She reported her ptosis was still evident, but improved, and that she had noticed her pupils were now consistently the same size. The neurologist had started the patient on low dose oral prednisone daily, oral hydrocodone for pain as needed, and instructed the patient to continue the 325mg of daily oral aspirin. Snellen visual acuities at this visit were 20/20 OD and 20/20 OS without correction. Her extraocular motilities were again full-range-of-motion with no signs of restriction OU. Confrontational visual fields were also again full-to-finger-count OU. The patient's anisocoric pupils at this time were improved and measured 3mm OD and 3mm OS in light conditions. Only a mild amount of anisocoria lingered in the dark which measured 6mm OD and 5mm OS. Neither a sign of APD nor light/near dissociation was found OU. Accommodative pupillary response was normal at this visit as well. Interpalpebral fissure width was improved and measured 11mm OD and 10mm OS. No signs of ipsilateral anhydrosis were found nor reported by the patient. Intraocular pressures measured 14mmHg OD and 12mmHg OS via Goldman tonometry. The remainder of the eye exam was stable and unchanged from previously. The patient was told to follow up with her PCP and neurologist as directed and was told to follow up at our clinic in 2–3 months for follow-up. A correspondence letter suggesting the use of intravenous heparin and/or oral warfarin was sent to the PCP in order to further prevent any ischemic events secondary to the underlying dissection, which has historically been the standard of care for ICAD's.
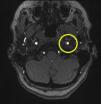
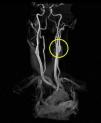
The patient returned for follow-up 3 months later. She reported the PCP and neurologist had, indeed, started her on oral warfarin based on our recommendations, and also continued her on the daily dose of 325mg aspirin to help prevent any ischemic attacks. The neurologist discontinued the hydrocodone and instead gave the patient oral gabapentin to manage her ongoing pain secondary to the ICAD. However, at the time of her 3 month follow-up visit with me she had since been instructed to discontinue her prednisone, warfarin, and gabapentin by her neurologist as her repeat MRI/MRA results from a few weeks prior had shown full resolution of her ICAD (see Figs. 5 and 6) and a drastic improvement in the caliber/lumen of the ICA. The patient was kept on 325mg daily aspirin by her PCP for prophylactic therapy. The patient reported her signs and symptoms had dramatically improved over the past few months and that she had not noticed any lingering ptosis or pupil abnormalities. She reported her neck pain and associated headaches had lessened in overall frequency as well. Snellen visual acuities at this 3-month follow-up appointment were stable at 20/20 OD and 20/20 OS without correction. Extraocular motilities were again full-range-of-motion with no signs of restriction OU. Confrontational visual fields were still full-to-finger-count OU. Pupils were equal, round, reactive to light OU with neither signs of APD OU nor light/near dissociation OU. Accommodative pupillary responses were normal OU as well. Pupils measured 5mm OD and 5mm OS in dark conditions and 3mm OD and 3mm OS in light conditions indicating resolution of her anisocoria. Interpalpebral fissure widths were equal in both eyes and measured 11mm OD and 11mm OS indicating resolution of the patient's ptosis. Intraocular pressures were measured at 16mmHg OD and 16mmHg OS via Goldman tonometry. The rest of the anterior segment and dilated fundus exams were completely unremarkable OU and consistent with her previous exams. The patient was instructed to follow up with her neurologist and PCP as directed for her ongoing care, and to follow up with our clinic in 6 months to monitor her ocular signs and symptoms.
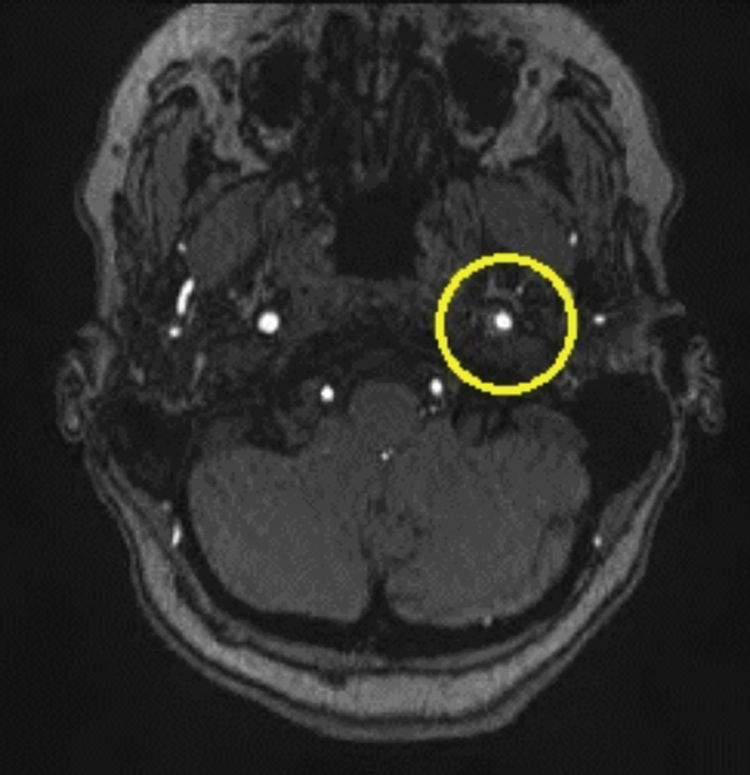
A thorough understanding of the oculosympathetic pathway is critical in understanding and detecting the underlying pathophysiology of Horner syndrome. The oculosympathetic pathway has a long and tortuous route from the hypothalamus prior to innervation of the ocular structures. It has three neuronal chains: first order, second order, and third order sympathetic chains. First order neurons originate in the hypothalamus and descend through the brainstem where it terminates at the approximate level of T1 known as the ciliospinal center of Budge–Waller. The second order neuron (preganglionic) exits the spinal cord, travels in the sympathetic chain over the apex of the lung, and ends approximately near the bifurcation of the common carotid artery located at the lower mandible of the jaw, at the superior cervical ganglion. The third order neuron (postganglionic) leaves the superior cervical ganglion, ascends as a plexus surrounding the internal carotid artery to the cavernous sinus, jumps off onto the abducens nerve (CN VI) for a short course, then onto the ophthalmic branch of the trigeminal nerve (CN V) where the fibers travel via the nasociliary nerve through the superior orbital fissure. The fibers then pass through, but do not synapse on the ciliary ganglion. Distal to the ciliary ganglion, the sympathetic fibers will continue in their course via the long posterior ciliary nerves to innervate the iris pupillary dilator muscle, Müller's muscle of the upper lid, and its unnamed companion muscle in the lower lid.2,4,5 The innervative fibers responsible for facial sweating travel on the external carotid artery therefore a dissection or insult to the internal carotid artery will result in only miosis and ptosis as facial sweating will not be impaired. Thus, an incomplete or partial Horner syndrome will occur with the absence of anhidrosis which was the case with our patient.2,5
ICAD is reported to have an incidence of 2.6–3.0 per 100,000.3,4 However, this may be falsely low as only two-thirds of patients will develop focal symptoms of cerebral or retinal ischemia prodding an ophthalmological or medical examination.3,4 Males and females have equal occurrence rates, although females tend to be 5 years younger at the time of dissection.13 While spontaneous ICAD account for only 2% of all ischemic strokes, it has been shown to be much more prevalent in the young to middle-aged populations, accounting for 10–25% of such cases.13 Average age of onset is reported to be 37.8–44 years in 70% of cases.3,4 Bilateral ICAD's are reported to occur only 20% of the time.3 Headache located ipsilaterally to the ICAD is the most common of all ICAD symptoms, occurring in 68–92% of patients.2,3,5,7,8 Intramural pain receptors in the ICA are assumed to be stimulated by the expanding hematoma which leads to the head/neck pain.8 Other symptoms/signs of ICAD most commonly include ipsilateral headache (68–92%), focal cerebral ischemia (49–67%), Horner syndrome (36–58%), subjective bruit (21–39%), and cranial nerve palsies (12–14%).2–5,7,8 The most common ocular signs/symptoms associated with an ICAD are: partial Horner syndrome (36–58%), amaurosis fugax (6–30%), ophthalmic/retinal arterial occlusion (5%), ischemic optic neuropathy (3.6%), and cranial nerve palsies (2.6%).2–5,7,8
ICAD are typically classified as either spontaneous or traumatic. Motor vehicle accidents are the most common cause of traumatic cases.2 Other traumatic etiologies, including trivial causes, that have been reported are: chiropractor neck adjustments, physical abuse, valsalva, sporting activities, exercise/resistance training, bar fights, strangulations, sexual intercourse, long phone calls holding phone between ear and the shoulder resulting in stretching of the neck, etc.3,13,14
The mechanism of action is best understood on a histological level of blood vessel anatomy. Typically, injuries causing an ICAD involve hyperextension and rotation of the neck.3 The internal carotid artery is stretched as it crosses the processes of the cervical vertebrae. The lamina intima, being the most inelastic layer of arterial vessels, tears under the forces of hyperextension and rotation. The blood in the ICA lumen then invades the space in the laminar wall creating a hematoma which typically dissects more distally, secondary to the direction of blood flow in the artery, than proximally, as well as for variable distances in the arterial wall.3 The intimal tear can thus result in ischemia, dissection, and/or thrombosis, etc.2,3
A history of hypertension, smoking, and oral contraceptive use have been shown to be more prevalent in ICAD patients, however atherosclerosis is distinctly uncommon in patients with ICAD.3,13 Some studies have shown an association between ICAD and pro-thrombotic states, as well as underlying connective-tissue systemic conditions such as: fibromuscular dysplasia, osteogenesis imperfecta, cystic medial necrosis, polycystic kidney disease, arteritis/vasculitis, Marfan's syndrome, and Ehlers–Danlos syndrome type IV.3,5,13,14 However, these causes are only identified in 1–5% of cases of ICAD.13,14 Of those that are identifiable, fibromuscular dysplasia and Ehlers–Danlos syndrome are the most common.13,14 However, in the vast majority of spontaneous ICAD's, the underlying pathogenesis remains unclear.3,13,14
As previously mentioned, Horner syndrome is the most common ocular finding in individuals with ICAD.2–5,7,8 Historically, pharmacologic testing has played an important role in diagnosing Horner syndrome. Cocaine solution with concentrations ranging from 2 to 5% can be used to confirm the diagnosis of Horner syndrome as cocaine blocks the re-uptake of norepinephrine in the nerve endings of the iris dilator muscles leading to dilation of the pupil.1 However, cocaine is a federally controlled drug and is difficult to obtain so its availability for use clinically is questionable for a wide range of practitioners.1 Higher concentrations (i.e. 10% cocaine solutions) have been associated with corneal epithelium compromise and are therefore not as commonly used.1 A normal pupil will dilate with cocaine solution (negative cocaine test), however a Horner's pupil will not (positive cocaine test).1 The maintenance of anisocoria after cocaine solution instillation diagnoses Horner syndrome, while dilation of both pupils after cocaine instillation occurs with physiologic anisocoria. Hydroxyamphetamine is the second drug used with Horner syndromes and has been used historically to localize the lesion after a positive cocaine test. If the pupil does not dilate with hydroxyamphetamine then the lesion is postganglionic. However, if dilation does occur then the lesion is assumed to be preganglionic in nature.1 This topical medication is available, however not widely distributed, therefore its use is limited in general practice as well. Also, cocaine and hydroxyampetamine must be separated by 24–72h in order to get accurate results as cocaine blocks the effectiveness of hydroxyamphetamine.1 This may lead to a delay in starting treatment which may not be in the patient's best overall interest.
A newer pharmacologic alternative has recently been suggested. There has been an increasing number of reports of both 0.5% and 1% apraclonidine (an alpha-two agonist with weak alpha-one receptor agonist affinity) being used in Horner syndrome patients for its diagnostic capabilities.9 It is believed that the use of 0.5–1% apraclonidine results in reversal of anisocoria secondary to denervation hypersensitivity of alpha-one receptors in the iris dilator muscle after 60min of drop installation.5,9,10 Also, lid ptosis was reported to improve in some patients with 0.5–1% apraclonidine as well.5,9,10 Therefore, the miotic pupil will dilate slightly with 0.5–1% apraclonidine leading to reversal of anisocoria.10 False negatives are possible with this method as it may take 5–8 days for denervation hypersensitivity to develop appropriately.10,11 Interestingly, the number of patients studied with apraclonidine in these studies is low, only 6 patients in one study and 8 in another, yet it appears to be a realistic diagnostic tool in acute presentations of oculosympathetic paresis.1,5,9,10 Cautious use of apraclonidine must be used in children due to side effects reported from its use in minors, particularly very young children, the most common side effects being lethargy and bradycardia.1,11 To simplify things, 0.5–1% apraclonidine has been shown to be a reliable diagnostic replacement of 2–5% cocaine solution as apraclonidine, like cocaine solution, is only used to diagnose and/or confirm a Horner syndrome, however hydroxyamphetamine and/or diagnostic imaging studies are still required to determine the actual location of the lesion. As a result of the above-mentioned pharmacologic side effect and availability issues, imaging studies of the sympathetic innervation of the ocular system have taken the forefront role in acute/acquired Horner syndrome workups, especially with a high suspicion of life-threatening underlying pathology such as ICAD.6
Historically, conventional angiography has been the gold standard for imaging ICAD's.2–5 However, side effects from the dye and risks of cerebral vascular incidents from the procedure itself have limited its use in today's era of diagnostic imaging studies. In recent years, less invasive imaging modalities such as computerized tomography (CT) including computerized tomography angiography (CTA) (80% sensitivity), magnetic resonance imaging (MRI) (78–84% sensitivity), and magnetic resonance angiography (MRA) have taken a forefront role in diagnosing the location of ICAD and/or other underlying causes of Horner syndrome.2,5 When MRI and MRA are used in conjunction to one another sensitivity reaches 95%.2,3
A recent 2011 study tried to investigate the proper protocol for imaging an acute/acquired Horner syndromes given the higher costs associated with imaging the entire oculosympathetic pathway and undetermined percentage yield of decisive underlying etiologies.12 However, given the low overall numbers of their study they concluded that a decisive yield and proper protocol for imaging of Horner syndromes could not be agreed upon at the current time and hence a separate study incorporating a larger study population would need to be undertaken in the future.12 Therefore, a clear and efficient diagnostic imaging protocol has not yet been proven and is left up to the attending physician at the time of the initial work-up. An additional topic that surfaces fairly frequently in the literature is that of imaging ICA's with duplex ultrasound given its lower cost. However, duplex ultrasound is typically not preferred in suspected cases of ICAD because of its decreased reliability/ability to detect and visualize dissections in the superior region of the neck, which is a common site of ICAD.2,3,5 However, duplex ultrasound has been shown to be a useful screening device as it can detect an abnormal blood flow pattern in more than 90% of patients, although it seldom visualizes the ICAD directly.13
Treatment for ICAD typically involves emergent anticoagulation/antiplatelet therapy followed by surgery and/or close observation via co-management with the patient's primary care physician, vascular surgeon, and/or neurologist.2–4 Anticoagulation is the standard of care in order to prevent any hemispheric and/or ocular vascular insults, which can have serious impacts on a patient's morbidity and/or mortality. While there has been no study to decisively determine which anticoagulant is the most beneficial, the consensus among clinicians/authors is one of urgent heparin followed by warfarin as the best treatment until the dissection resolves on its own over time.2,4 Anticoagulation with an internal normalized ratio (INR) of 2.0–3.0 is normally used for 3–6 months.13 In patients who have no ischemic symptoms, antiplatelet therapy alone may be all that is indicated.13 ICAD resolution usually occurs within a 3–6 month time period, at which time treatment can be discontinued once imaging studies have proven the dissection to be fully resolved and lumenal patency restored.2,3,13 If anticoagulant use fails or is contraindicated, then either close monitoring or surgery via intravascular stents are considered which can immediately restore lumenal diameter.2,3,13 Intravascular stents are the surgical treatment of choice if medical treatment fails.13
Overall ICAD prognoses are very good as 85–90% of patients will have complete resolution in 3–6 months with appropriate therapy, and the associated headaches typically resolve in 95% of individuals during this time span as well.2–4 Any lingering and stable ptosis after ICAD resolution can typically be repaired via blepharoplasty for those patients with cosmetic concerns.6 Recurrent ICADs are considered rare at best but the risk is higher in those with a heritable arteriopathy.13 In unaffected arteries, ICAD recurrence rates are estimated to be 2% during the first month then decreases to 1% per year thereafter.13 Recurrence of ICAD in the same artery is considered very rare.14 Although death is certainly a possibility, mortality rarely exceeds 5%.3,13,14
ConclusionIn our patient's case, in-office pharmacologic testing was deferred given the obvious diagnosis of an acute-onset Horner syndrome based upon the patient's history, presenting signs, and her painful symptoms which necessitated an urgent diagnostic investigation. Given the acute and rapid onset of her signs and symptoms, the lack of time necessary for denervation hypersensitivity to appropriately develop would have made apraclonidine or additional pharmacologic testing difficult to rely on.
The patient's Horner syndrome, as discussed previously, was caused by an ICAD most likely associated with her history of running a short race a few days earlier. The process of her stopping mid-race to bend down and tie her shoe may have set off her cascade of signs and symptoms. As she bent down, her neck most likely hyperextended and rotated to some degree, which stretched the lamina intima layer of the carotid artery leading to a small defect in the arterial wall. In conjunction to this, her blood pressure would have been elevated if she was exercising/running even mildly. Therefore the higher pressure and flow of blood would surge into the “opening/defect” in the arterial wall leading to the dissection worsening distal to the origin site as she continued to finish the race. In support of this, a literature review clearly supports cases of ICAD secondary to trivial trauma and sporting events such as in this case.3,13,14
It is difficult to determine if she, indeed, had an underlying inherent structural defect in the wall of her carotid arteries, secondary to such systemic conditions as previously mentioned in this article. If she did, this would have increased her risk of ICAD which may have been aggravated by her short marathon race and bending over to tie her shoes. A letter requesting a work-up to rule out pro-thrombotic states, fibromuscular dysplasia, osteogenesis imperfecta, cystic medial necrosis, Marfan's syndrome, arteritis/vasculitis, and Ehlers–Danlos syndrome was sent to the patient's primary care physician (PCP). Although, one would think that spontaneous ICAD does not occur unless there is a concurrent underlying systemic disorder, the literature shows that “in the majority of patients, the pathogenesis is unclear”.3,13 Unfortunately, at the time of writing this review, the patient's PCP had not pursued any further testing for underlying connective tissue disorders and/or other possible underlying systemic conditions that have been associated with ICAD despite my recommendations otherwise. Nonetheless, she is being watched closely by our clinic, her PCP, and her neurologist. Her long-term prognosis for a full recovery with no restrictions or residual complications from her ICAD is very good given her young age and healthy lifestyle in general. She will continue to be followed closely until her signs and symptoms have proven consistently stable in nature.
Horner syndrome secondary to an ICAD is a rare, but well-reported finding in ophthalmological literature. Timely recognition and referral for urgent imaging studies and anticoagulation medications with the patient's primary care physician are important for eye care providers in order to help prevent any ischemic insults to the brain and/or ocular tissues that can affect morbidity and/or mortality of patients. Appropriate management of these patients typically results in very good prognoses and outcomes for the vast majority.
In cases of acute, painful Horner syndrome without a clear preceding etiology and high clinical index of suspicion for ICAD, the author recommends emergent CTA/MRA of the carotid artery system to be seriously considered by all physicians involved in the management of the case. Emergent MRI of the entire oculosympathetic pathway should also be considered in acute Horner sydnrome without clear etiology to rule out other potential ominous causes, in addition to ICAD. Likewise, underlying systemic conditions should be investigated in all patients with ICAD via the patient's primary care provider to rule out any further inherent structural defects or other potential complications which may be managed appropriately to prevent any possible future medical complications.
Conflict of interestsThe author has no financial or other relationships that might lead to conflict of interest.
Special thanks to Iowa Methodist Hospital's Radiology unit and Charles DePena, M.D. for their help in interpreting and highlighting the radiologic scans involved with this case.





