

Dementia, including Alzheimer's disease (AD), is a major disorder causing visual problems in the elderly population. The pathology of AD includes the deposition in the brain of abnormal aggregates of β-amyloid (Aβ) in the form of senile plaques (SP) and abnormally phosphorylated tau in the form of neurofibrillary tangles (NFT). A variety of visual problems have been reported in patients with AD including loss of visual acuity (VA), colour vision and visual fields; changes in pupillary response to mydriatics, defects in fixation and in smooth and saccadic eye movements; changes in contrast sensitivity and in visual evoked potentials (VEP); and disturbances of complex visual functions such as reading, visuospatial function, and in the naming and identification of objects. Many of these changes are controversial with conflicting data in the literature and no ocular or visual feature can be regarded as particularly diagnostic of AD. In addition, some pathological changes have been observed to affect the eye, visual pathway, and visual cortex in AD. The optometrist has a role in helping a patient with AD, if it is believed that signs and symptoms of the disease are present, so as to optimize visual function and improve the quality of life.
Las distintas formas de demencia, entre las que se incluye la enfermedad de Alzheimer (EA), son graves trastornos que causan problemas visuales en las personas de edad avanzada. La patología de la EA incluye la acumulación en el cerebro del péptido β-amiloide (Aβ en forma de placas seniles (PS) y de proteína tau anormalmente fosforilada en forma de ovillos neurofibrilares (ONF). Se ha documentado un amplio espectro de problemas visuales en pacientes con EA, entre los que se incluye la pérdida de agudeza visual (AV), problemas relacionados con la visión de color y el campo visual, cambios en la respuesta pupilar ante midriáticos, problemas de fijación así como movimientos oculares sacádicos y de seguimiento anormales; cambios en la sensibilidad al contraste y en los potenciales evocados visuales (PEV); y alteraciones de algunas funciones visuales complejas, tales como la lectura, la función visuoespacial, y a la hora de nombrar y de identificar objetos. Muchos de estos cambios no están exentos de cierta controversia, puesto que en la literatura aparecen datos contradictorios al respecto y, además, no hay ninguna característica visual u ocular en la que se pueda basar el diagnóstico de la EA. Además, se ha observado que algunos de los cambios patológicos que aparecen en la EA afectan al ojo, a la vía óptica y al córtex visual. El optometrista juega un papel destacado a la hora de ayudar a los pacientes que padecen EA. Si se sospecha que existen signos y síntomas de la enfermedad, la ayuda en la optimización de la función visual contribuirá para mejorar su calidad de vida.
A significant part of current optometric practice is directed to the visual problems of the elderly and this trend is likely to continue, given the demographic increase in the geriatric community worldwide. A major disorder causing visual problems in the older population is dementia and especially Alzheimer's disease (AD), which is the commonest form of dementia to affect the elderly.1
AD is a degenerative disorder of the nervous system affecting approximately 10% of individuals aged 65 or over. It is estimated that 24.3 million individuals worldwide have dementia, with 4.6 million new cases recorded every year.2 There have been various estimates of the prevalence of AD in the elderly population.2,3 With advancing age, the prevalence of the disease increases to an estimated 19% in individuals aged 75-841, and is 30-35% for those older than 85 years.2 The development of dementia involves a decline in short-term memory, impairment of judgment, and a loss of emotional control. These symptoms begin insidiously, develop slowly over a period of years, and result ultimately in the complete breakdown of the mental function. A small proportion of cases of AD (<5%) have a genetic origin, but most cases occur sporadically within the population.4
AD is a clinical diagnosis and during the early stages of the disease it can be difficult to distinguish it from other forms of dementia such as Pick's disease (PiD) or dementia with Lewy bodies (DLB), as well as from dementias associated with a known specific cause, such as normal pressure hydrocephalus, vascular disease, viral infection, or exposure to drugs. Despite the difficulties, however, the final success rate in the clinical diagnosis of AD is usually quoted to be 75-90%.5 The clinical diagnosis of AD is based on criteria developed originally by the ‘National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association’ (NINCDS-ADRDA) work group6 and modified by the National Institute on Aging (NIA) - Reagan Institute.7 The definitive diagnosis of AD, however, requires examination of brain tissue either by means of a biopsy or post-mortem. The pathology of AD includes the deposition in the brain of abnormal aggregates of β-amyloid (Aβ) in the form of senile plaques (SP) and abnormally phosphorylated tau in the form of neurofibrillary tangles (NFT) (Figure 1). Significant densities of SP and NFT in the cerebral cortex of the brain are required for a pathological diagnosis of AD. There are no treatments that can arrest the progression of AD, but some drugs can successfully treat the symptoms for a period of time.
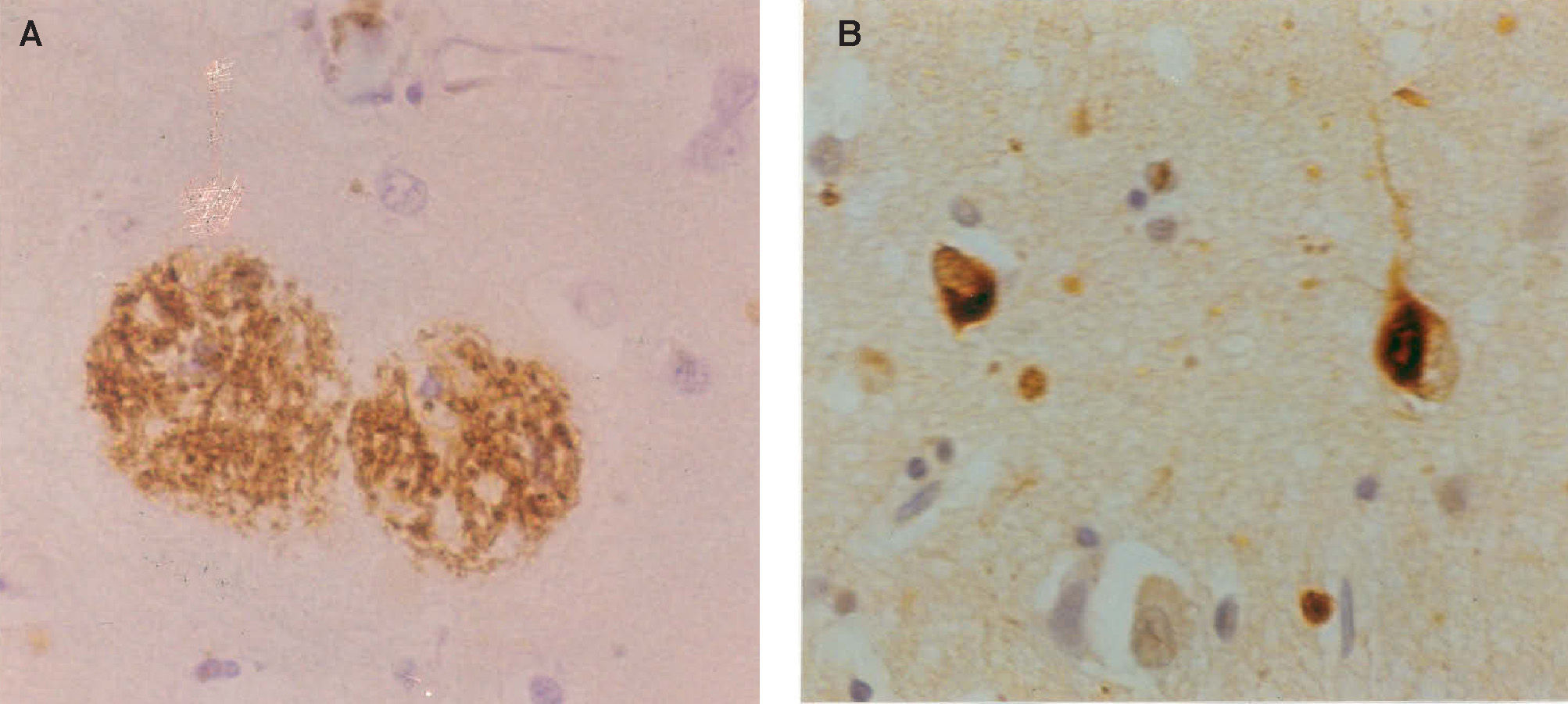
 deposits in the form of senile plaques (SP) in a section of the cerebral cortex. Deposits appear as brown patches and are widely distributed, especially in the cerebral cortex (β-amyloid immunohistochemistry), B. neurofibrillary tangles (NFT) in the cerebral cortex appearing as inclusion bodies within neurons (tau immunohistochemistry).")
Neuropathology of Alzheimer's disease: A. β-amyloid (Aβ) deposits in the form of senile plaques (SP) in a section of the cerebral cortex. Deposits appear as brown patches and are widely distributed, especially in the cerebral cortex (β-amyloid immunohistochemistry), B. neurofibrillary tangles (NFT) in the cerebral cortex appearing as inclusion bodies within neurons (tau immunohistochemistry).
AD can also affect the eye, visual pathway, and visual cortex and result in various visual signs and symptoms. Because of the increasing numbers of elderly people in the population, the optometrist is likely to see elderly patients in their practice, some of whom will have or will be developing AD, and is expected to have a role in managing their visual symptoms.8 Hence, this review describes the general pathology of AD, the ocular and visual features that have been described, and the pathological changes in the visual system that may explain these symptoms. The literature search began with several of the major reviews of the topic dating from 1989-19967,9-12 and then proceeded to examine specific changes in AD from 1996 to the present as compiled by PUBMED and ISI Web of knowledge.
Pathology of Alzheimer's Disease (AD)General FeaturesAD is characterized by a large number of pathological changes affecting the brain. Few of these changes are specific to AD, however, and many appear to be exacerbations of normal aging.13 There is atrophy of the cerebral hemispheres with narrowed gyri and widened sulci and widening of the subarachnoid space.14 These gross changes mainly affect the frontal, temporal, and parietal lobes and often spare the occipital lobe. The meninges are thickened by fibrosis and the ventricles are often dilated.15 The white matter appears yellow and ‘rubbery’ and there is ‘spongiosis’ (development of vacuolation) and ‘gliosis’ (a proliferation of glial cells which is often associated with pathological changes in the brain) affecting the grey matter.15 These changes may be more apparent in cases of early onset (<65 years) while some late-onset cases (>65 years) may exhibit little atrophy.16
Histological FeaturesEver since the first descriptions of pre-senile dementia by Alois Alzheimer in 1907,17 the formation of SP and NFT (Figure 1) have been regarded as the defining histological features of Alzheimer's disease (AD).7,18,19 AD formally became recognized as a disease entity in 1910 and was named after Alzheimer by Kraepelin, based on the clinical and pathological description of the original cases. The most important molecular constituent of the SP is β-amyloid (Aβ)20 and, consequently, SP are also referred to as Aβ deposits. Several types of Aβ deposits have been identified in AD brains, but the majority can be classified into three morphological subtypes:21,22 1) diffuse deposits, in which most of the Aβ peptide is not aggregated into fibrils and dystrophic neurites and paired helical filaments (PHF) are infrequent or absent, 2) primitive deposits, in which the Aβ is aggregated into amyloid and dystrophic neurites and PHF are present, and 3) classic deposits, in which Aβ is highly aggregated to form a central ‘core’ surrounded by a ‘ring’ of dystrophic neurites. The latter type of deposit is especially common in the visual cortex.23,24
The most important molecular constituent of the NFT is the microtubule-associated protein (MAP) tau, which is involved in the assembly and stabilization of the microtubules and, therefore, establishes and maintains neuronal morphology.25,26 In normal neurons, tau is soluble and binds reversibly to microtubules with a rapid turnover.27 In AD, however, tau does not bind to the microtubules but collects as insoluble aggregates to form PHF which resist proteolysis and accumulate as NFT. Tau extracted from AD brains consists of both soluble and insoluble forms where, in the latter case, the tau present is in an abnormally phosphorylated isoform.28
Pathogenesis of ADStudies of the molecular composition of Aβ deposits and NFT have played an important role in the development of hypotheses as to the pathogenesis of AD. For example, the discovery of Aβ as the most important molecular constituent of the SP20 led to the development of the ‘Amyloid Cascade Hypothesis’ (ACH), the most important model of the molecular pathology of AD developed to date.29 The ACH proposes that the deposition of Aβ is the initial pathological event in the disease, leading to the formation of NFT, cell death, and, ultimately, dementia. A number of gene mutations have been implicated in familial forms of AD. Rare cases are linked to mutations of the amyloid precursor protein (APP) gene on chromosome 2130,31 and a larger group to the presenilin (PSEN) genes PSEN1 on chromosome 1432 and PSEN2 on chromosome 1.33 In addition, the apolipoprotein E (apo E) gene on chromosome 19 is an important risk factor associated with late-onset familial and sporadic AD,34 possession of one or more E4 alleles significantly increasing the risk of developing the disease.
There are, however, observations that are difficult to reconcile with the ACH. First, in transgenic mice, genes over-expressing the amyloid precursor protein (APP) do not produce the predicted series of pathological events.35,36 Second, SP and NFT appear to be separated in the brain both temporally37 and spatially,38 suggesting that the two lesions may develop independently. Indeed, in the entorhinal cortex, the NFT may actually precede the appearance of SP.36 The uncertainty as to the significance of SP and NFT in AD has led to alternative models being proposed to explain the aetiology of AD based on perturbation of vesicular trafficking at synapses, disruption of the cytoskeletal network, or the distribution of membrane cholesterol.39
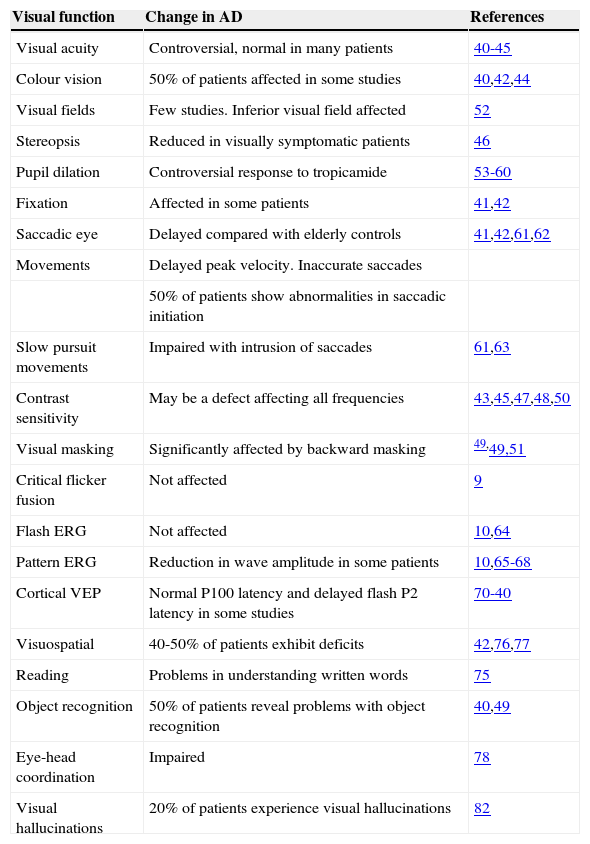
Visual Changes in Alzheimer's DiseaseA number of visual changes have been reported in AD patients and these are summarized in table 1.
Functional changes in vision in Alzheimer's disease
| Visual function | Change in AD | References |
| Visual acuity | Controversial, normal in many patients | 40-45 |
| Colour vision | 50% of patients affected in some studies | 40,42,44 |
| Visual fields | Few studies. Inferior visual field affected | 52 |
| Stereopsis | Reduced in visually symptomatic patients | 46 |
| Pupil dilation | Controversial response to tropicamide | 53-60 |
| Fixation | Affected in some patients | 41,42 |
| Saccadic eye | Delayed compared with elderly controls | 41,42,61,62 |
| Movements | Delayed peak velocity. Inaccurate saccades | |
| 50% of patients show abnormalities in saccadic initiation | ||
| Slow pursuit movements | Impaired with intrusion of saccades | 61,63 |
| Contrast sensitivity | May be a defect affecting all frequencies | 43,45,47,48,50 |
| Visual masking | Significantly affected by backward masking | 49,49,51 |
| Critical flicker fusion | Not affected | 9 |
| Flash ERG | Not affected | 10,64 |
| Pattern ERG | Reduction in wave amplitude in some patients | 10,65-68 |
| Cortical VEP | Normal P100 latency and delayed flash P2 latency in some studies | 70-40 |
| Visuospatial | 40-50% of patients exhibit deficits | 42,76,77 |
| Reading | Problems in understanding written words | 75 |
| Object recognition | 50% of patients reveal problems with object recognition | 40,49 |
| Eye-head coordination | Impaired | 78 |
| Visual hallucinations | 20% of patients experience visual hallucinations | 82 |
Visual acuity (VA) can be difficult to measure accurately in patients with AD during the later stages of the disease, but studies suggest that VA is normal in the early stages of the disease.40-44 Low-contrast acuity using Regan charts presented at four contrast levels, however, shows a reduction in acuity with contrast in AD.45 In patients in whom impairments of VA have already been demonstrated, there may be impairments in stereopsis as measured by random dot stereograms.46
Colour VisionWhether or not there is defective colour vision in AD is controversial, with some studies suggesting normal colour vision in patients with mild to moderate AD.44 In other studies, however, defective colour vision may be present in approximately 50% of patients.40,42 There is little available information on whether there are specific colour vision problems in AD; e.g., affecting the Red/Green (R/G) rather than Blue/Yellow (B/Y) axis. The ability to use colour information may be impaired in AD. Hence, in cognitive tasks in which colour is used as an attention enhancer, a cue, or as a distracter, patients with AD were less accurate in their performance than controls.44
Contrast SensitivityVA provides information about the ability of the patient to resolve higher-contrast spatial details. The contrast sensitivity function (CSF), however, provides a measure of the performance across a wider range of spatial frequencies and contrasts. The performance of patients with AD is not clear. Some studies have not reported changes in contrast sensitivity in AD.43 Other studies have reported reduced contrast sensitivities in AD over all spatial frequencies,45,47,48 and others at lower spatial frequencies only.49 Differences in reported performance may be attributable to variation in patient population or assessment methods and especially failure to account for VA differences between groups.50 Hence, it is likely that there are contrast sensitivity changes in AD, but it is not possible to be specific about the frequencies affected.
Critical Flicker Fusion Frequency Threshold and Visual MaskingThe critical flicker fusion threshold, i.e., the lowest frequency at which the patient can no longer perceive a flickering light as a steady light, is normal in AD.9 Patients with AD often show deficits on visual masking tasks.49 Visual masking involves the presentation of a second stimulus immediately before (forward masking) or after (backward masking) a test stimulus. Patients with AD are significantly affected by a backward patterned mask stimulus compared with age-matched controls.51 This result suggests that the speed of central visual processing, which often decreases with age, is reduced even further in AD.
Visual FieldsThere have been few studies of the visual fields in AD. In one report, visual sensitivity was reduced throughout the visual field but deficits were most pronounced in the inferior field.52 In addition, in follow up studies of patients with AD, it was found that there may be a progression of visual field loss over time.52
Pupillary FunctionThere has been considerable interest in whether patients with AD exhibit an abnormal pupillary response to the muscarinic receptor antagonist tropicamide, widely used by optometrists as a mydriatic.53 Early reports suggested that some patients with AD display a specific response to low doses (typically 0.01%) of tropicamide, with pupils dilating at least 13% more compared with normal elderly controls.35 However, subsequent studies suggest that the tropicamide test is not a reliable method of diagnosing AD. For example, there is no difference between the response of AD patients and that of patients with vascular dementia (VaD)55 or Parkinson's disease.56 In addition, not all studies have shown a significantly different response between AD and elderly control patients although a difference was demonstrated compared with young controls.55 Hence, use of the tropicamide response cannot be recommended as a clinical application to detect AD.57,58
Enhanced pupillary responses in AD have also been reported following the application of dilute solutions of phenylephrine (a sympathetic agonist) and of pilocarpine (a cholinergic agonist).59 Reductions in the pupillary light reflex have also been reported in AD, although these responses are highly variable.60
Eye MovementsThe ability of some patients to fixate a target is affected in AD.41,42 Defects of fixation control may be associated with degeneration of the parietal lobe of the brain, a region believed to be involved in maintaining fixation stability. Several changes in saccadic eye movements have been reported. First, saccadic latency declines with age but the delays become more pronounced in patients with AD.61 Second, saccadic velocities are reduced, the degree of this reduction being correlated with the severity of the dementia. Third, patients at more advanced stages of the disease exhibit inaccurate saccades with undershooting of the target by 10-30%.62 Fourth, 50% of the patients with AD have difficulty in initiating or maintaining saccadic eye movements.62
Smooth pursuit eye movements, which are a sensitive indicator of brain function, may also be affected.63 A gradual deterioration of these movements occurs in AD with catch up saccades being necessary to maintain fixation.61 Degeneration and atrophy of the frontal and/or the parietal lobes may be responsible for these changes.
ElectrophysiologyA number of electrophysiological changes have been recorded in patients with AD. First, changes in the electroretinogram (ERG) have been reported in some patients but not in others. For example, the amplitude of the pattern-ERG (PERG) response is reduced, although the flash electroretinogram ERG may be unaffected.10,64 A significant delay in the N35, P50, and N95 components of the PERG, together with reductions in amplitude have been reported accompanied by significant reductions in nerve fibre layer thickness.65 In other studies the amplitude and latency of the components of the PERG have been reported to be unaffected.66-68 The reduction in the ‘b’ wave shown in some studies could be attributable to the reduced number of ganglion cells in the retina in AD patients.65,69
Second, a number of studies dating from the 1980s have suggested that the latency of the flash P2 component of the cortical visual evoked response (VEP) is delayed and the P100 component to a reversing checkerboard is normal in patients with AD.70-73 This pattern of abnormalities, however, has not been shown in all studies and has not been adopted subsequently as a routine test for AD.74
Complex Visual FunctionsPatients with AD exhibit difficulties with more complex visual functions such as reading75, visuospatial function42,76,77 and in the identification and naming of objects.40 Significantly greater thresholds for perceiving shapes defined by motion cues49 may be present and this is likely to affect object recognition. Patients with AD also show impairment of eye-head coordination,78 problems with finding objects when surrounded by others,79 and in finding known objects in an unknown environment.80 Deficient perception and cognition in AD are often attributed to slow information processing. Increasing ‘stimulus strength’ by for example increasing stimulus contrast can often improve various aspects of the cognitive performance in AD.81
In rare cases, patients develop a particular combination of visual symptoms called ‘Balint's syndrome’ before any signs of dementia are apparent.82 These include a psychic paralysis of gaze (ocular apraxia), optic ataxia (lack of muscular coordination) e.g., an inability to guide the hand towards an object using visual information, and a spatial disorder of attention (‘simultanagnosia’): viz., an inability to report all items or their relationships in pictures depicting events or situations.11 These symptoms are accompanied by visual field constriction, the fading of centrally fixated objects, and impaired reading ability despite normal VA. It is possible that these symptoms are attributable to degeneration of more complex visual areas rather than to retinal dysfunction or to problems affecting the primary visual pathway.11
Visual HallucinationsVisual hallucinations may be present in some patients, especially in those with impaired VA and with more severe cognitive impairment.82 Visual hallucinations, however, may accompany many disorders including Creutzfeldt-Jakob disease and DLB.82
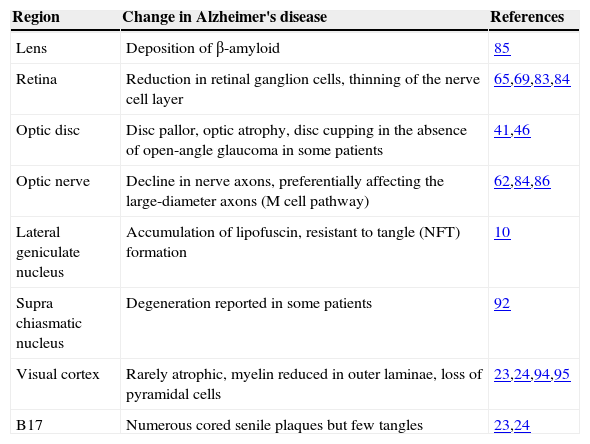
Pathological Changes in the Visual SystemPathological changes that have been observed in the visual system in AD are summarised in table 2. Pathological changes may occur at several sites in the visual system from eye to brain, and hence, the visual signs and symptoms observed in AD are likely to reflect a combination of both ocular and neural factors.
Pathological changes in the visual system in Alzheimer's disease
| Region | Change in Alzheimer's disease | References |
| Lens | Deposition of β-amyloid | 85 |
| Retina | Reduction in retinal ganglion cells, thinning of the nerve cell layer | 65,69,83,84 |
| Optic disc | Disc pallor, optic atrophy, disc cupping in the absence of open-angle glaucoma in some patients | 41,46 |
| Optic nerve | Decline in nerve axons, preferentially affecting the large-diameter axons (M cell pathway) | 62,84,86 |
| Lateral geniculate nucleus | Accumulation of lipofuscin, resistant to tangle (NFT) formation | 10 |
| Supra chiasmatic nucleus | Degeneration reported in some patients | 92 |
| Visual cortex | Rarely atrophic, myelin reduced in outer laminae, loss of pyramidal cells | 23,24,94,95 |
| B17 | Numerous cored senile plaques but few tangles | 23,24 |
Whether or not patients with AD show characteristic pathological changes within the eye, observable during fundus investigation, is controversial. Some studies have failed to detect any fundus abnormalities in AD except those to be expected in normal aged individuals.46 Nevertheless, a proportion of patients may show abnormalities including disc pallor, optic atrophy, and disc cupping.41
Retinal nerve fibre layer abnormalities have been studied in AD and in age-matched controls using retinal photography. A higher proportion of patients with AD exhibit abnormalities in these tests compared with controls, but there is often disagreement between observers as regards the interpretation of photographs, especially in more advanced cases of AD.83 Hence, it is possible that there is ganglion cell degeneration in AD. Consistent with this suggestion, significant reductions in the retinal nerve fibre layer thickness using optical coherence tomography have also been shown in a group of 17 AD cases.65
Studies of the retinae of patients with AD obtained postmortem suggest a reduction in the number of ganglion cells and in the thickness of the nerve cell layer.69 Ganglion cells may also be swollen or shrunken and some may contain vacuoles. A reactive gliosis may accompany these changes. A decline in the number of retinal ganglion cells could explain the disc abnormalities observed in some patients.84 Openangle glaucoma was unlikely to be the cause in these cases, since intraocular pressure was recorded to be less than 19mm Hg in all patients, and none of the patients with AD had a family history of glaucoma.84 Nevertheless, low-tension glaucoma cannot be ruled out in these patients. In addition, Aβ deposits may also be found in the lens of the eye in patients with AD.85
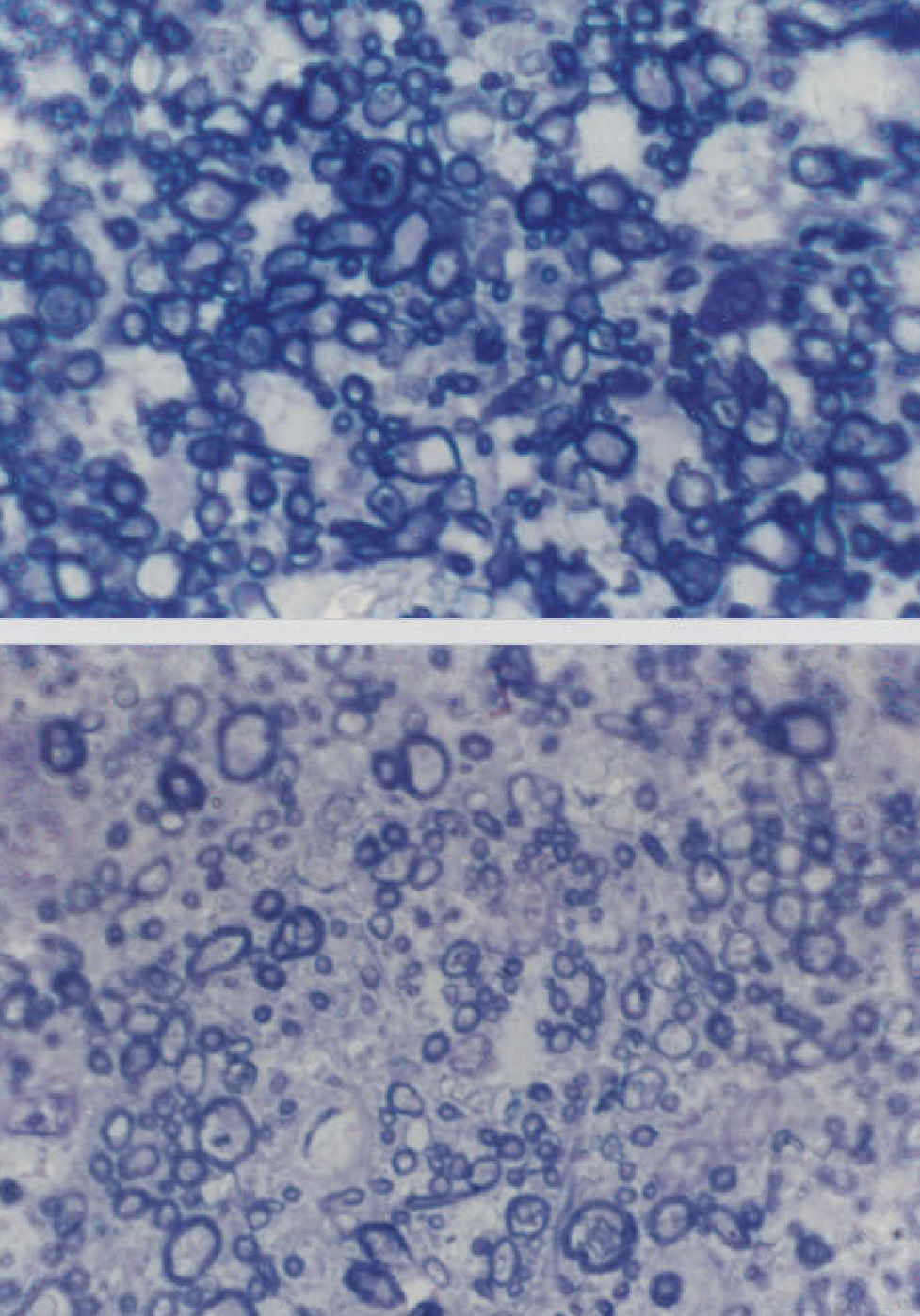
Changes Affecting the Visual PathwayA well established pathological change in patients with AD is the decline in the density of optic nerve fibres (Figure 2).62,84 In some studies, a preferential decline in the largerdiameter axons has been reported,62,84 whereas in other studies a decline in the smaller-diameter axons may be present.86 There may be subtypes of AD in which degeneration affects preferentially either the large or small axons. Whether this degeneration reflects a decline in retinal ganglion cells, degeneration of the visual cortex affecting the retina as a consequence of retrograde degeneration, or both is yet to be established.
 (sections stained with toluidine blue, Magnification bar represents 5 μm).")
Cross-sections of part of the optic nerve showing the axon profiles. The upper picture shows axon profiles corresponding to an elderly control patient and the lower picture corresponds to a patient with Alzheimer's disease (AD) (sections stained with toluidine blue, Magnification bar represents 5 μm).
A decline in large-diameter axons in the optic nerve suggests that it is the M- rather than the P-cell pathway that is affected in AD. This pathway, which is stimulated using low contrast and luminance stimuli and by coarse patterns is essentially a ‘luminance’ channel involved in motion detection.87 Its specific degeneration could explain the abnormalities in the cortical flash VEP that have been reported previously in some studies. Hence, some studies have suggested that the latency of the flash P2 component is delayed while the P100 component to a reversing checkerboard stimulus is normal in patients with AD.88-90 However, as mentioned above, critical flicker fusion frequency threshold is apparently normal in AD9 and this is also mediated by M cells. If the smaller-sized axons are affected in the optic nerve of AD patients, then the data suggest impairment of the P pathway. This pathway is more significantly involved in the detection of fine details of the visual scene and with colour vision.87 Consistent with the involvement of the P pathway, abnormalities of colour vision have been observed in up to 50% of patients with AD.42
Degeneration of the lateral geniculate nucleus (LGN) is rare in AD.10 Neurons in this region are some of the most resistant in the nervous system to the development of cellular NFT. However, cells in the LGN accumulate lipofuscin, a pigment found in increasing amounts in nerve cells with age.10
Suprachiasmatic Nucleus (SCN)The suprachiasmatic nucleus (SCN) is reported to degenerate in some Alzheimer's patients.91 This structure, located within the hypothalamus, receives an input from the retina and is involved in regulating the timing of sleep. Hence, degeneration of the SCN may explain the sleep disturbances reported in many AD patients.92
Pathology of the Visual CortexAlthough atrophy of the parietal lobe of the brain is common in AD, this rarely spreads to involve all of the occipital lobe. Pathological changes within the visual cortex normally involve the visual association areas and spare to some degree, the primary visual cortex (area V1). However, a number of pathological changes have been reported in the visual cortex in Alzheimer patients. In a retrospective study of 106 patients with AD,24 SP and NFT were observed in the visual cortex in 72% and 27% of cases respectively. The density of SP and especially NFT is generally greater in the visual association areas (V2, V3 etc.) than in V1 especially in younger cases. In area V1, SP with distinct amyloid cores and relatively few NFT can be observed while in V2, numerous uncored ‘neuritic’ plaques and tangles are usually present.23 Whether differences in cortical pathology between areas V1 and V2 are responsible for the cortical VEP to flash and pattern stimuli described previously remains to be established.93 In a significant proportion of cases of AD, the density of SP and/or NFT is significantly greater in the cuneal compared with the lingual gyrus of area V1. This difference could contribute to the predominantly inferior visual field deficits reported in one study.52 In addition, the amount of myelin appears to be reduced in the outer laminae of the visual cortex and loss of neurons and neurotransmitters have been reported.23
The Optometrists RoleAs primary eye-care practitioners, optometrists should be able to identify the visual problems of patients with AD and should be expected to work with patients and their carers to manage their visual welfare.95
In the Identification of ADThe optometrist has a role in helping a patient with AD if it is believed that signs and symptoms of the disease are present and especially if no diagnosis has yet been made. The symptoms of AD are highly variable; some patients not exhibiting any visual symptoms while others may show various combinations of such symptoms. In addition, the literature regarding the visual changes in AD is controversial, with different studies often giving conflicting results. In addition, optometrists should not use the 0.01% tropicamide test as a test for AD, as recent studies57,58 have not confirmed earlier reports.53 However, it is important that optometrists continue to use the regular dosage of tropicamide (1%) for papillary dilation and ocular health examination.
Detecting the Visual Problems of the PatientThe following tests and procedures may be useful in identifying the visual problems of a patient with AD. It is particularly important to carry out the full examination, including ocular health assessment. Subsequently, several additional tests may be helpful such as regular assessment of binocular vision. Hence, the ‘motility test’ should be carried out and the practitioner should notice whether the movement of the eyes is smooth and whether or not they fall behind the target. In addition, saccadic eye movements could be tested. The patient should be asked to fixate an object in the primary position as fixation is affected in a proportion of patients, the patient's eyes often drifting away from the target.41,42 The patient could be asked to read a page of print since a patient with AD is likely to have problems with this task and to exhibit a reduction in reading speed compared with normal subjects. Finally, colour vision could be tested monocularly and binocularly as a defect may be present in as many as 50% of patients.40,41
Practical Ways the Optometrist Can HelpPatients with AD are likely to have visual problems that have not been detected due to the developing dementia. The dementia population generally is less likely to be able to describe their visual problems effectively and is more likely to experience and tolerate visual deficits.96 In a sample of 85 patients with AD in a nursing home environment, for example, 80 required spectacles for the correction of presbyopia, myopia or both.96 Of these patients, 25 had not actively been using spectacles since entering the nursing home; nine were too cognitively impaired to request them, eight had lost or damaged their spectacles, and eight had prescriptions no longer accurate enough to correct their vision. One factor that improves the quality of life of a patient with AD and that, as a consequence, reduces the burden on those that care for the patient, is for the patient to be able to see as clearly as possible. Hence, it is important that the visual problems present should be investigated and detected by the eye practitioner and those due to ocular factors corrected as far as possible. In addition, it is important to bring to the attention of carers visual problems of the patient that cannot be corrected, such as oculomotor apraxia, field defects, or problems with colour vision. As a consequence of such information, carers can make accommodations for these problems, e.g., by careful use of colours or in the positioning of objects.
Financial disclosure: There are no financial interests relating to this review.





