

In recent years, the term mitochondrial optic neuropathy (MON) has increasingly been used within the literature to describe a group of optic neuropathies that exhibit mitochondrial dysfunction in retinal ganglion cells (RGCs). Interestingly, MONs include genetic aetiologies, such as Leber hereditary optic neuropathy (LHON) and dominant optic atrophy (DOA), as well as acquired aetiologies resulting from drugs, nutritional deficiencies, and mixed aetiologies. Regardless of an inherited or acquired cause, patients exhibit the same clinical manifestations with selective loss of the RGCs due to mitochondrial dysfunction. Various novel therapies are being explored to reverse or limit damage to the RGCs. Here we review the pathophysiology, clinical manifestations, differential diagnosis, current treatment, and promising therapeutic targets of MON.
En los últimos años, se ha incrementado el uso en la literatura del término neuropatía óptica mitocondrial (MON), para describir un grupo de neuropatías ópticas que presentan una disfunción mitocondrial en las células ganglionares de la retina (RGC). De manera interesante, las MON incluyen etiologías genéticas, tales como Neuropatía Óptica Hereditaria de Leber (LHON) y Atrofia Óptica Dominante (DOA), así como etiologías adquiridas derivadas del consumo de drogas, deficiencias nutricionales y etiologías mixtas. Independientemente de que la causa sea hereditaria o adquirida, los pacientes presentan las mismas manifestaciones clínicas, con pérdida selectiva de RGCs debido a la disfunción mitocondrial. Se están explorando diversas terapias novedosas para revertir o limitar el daño a las RGC. En este documento revisamos la patofisiología, las manifestaciones clínicas, los diagnósticos diferenciales, el tratamiento actual y los prometedores objetivos terapéuticos de las MON.
With advances in the understanding of optic nerve diseases, a new term has emerged within the literature to describe a group of diseases caused by mitochondrial dysfunction of the optic nerve: mitochondrial optic neuropathy (MON).
MON describes a group of optic neuropathies with similar clinical features and the same pathophysiology of mitochondrial dysfunction. Regardless of the underlying inherited or acquired cause, MONs share the common feature of mitochondrial dysfunction in the prelaminar area of the optic nerve. The subsequent selective damage and loss of retinal ganglion cells (RGCs) result in clinical signs of painless, progressive bilateral visual acuity loss, central or cecocentral scotomas, and color vision deficiencies. Although the same clinical features exist in MONs, the prevalence of specific aetiologies varies. In acquired MONs, the disease has no gender, racial, or age prevalence. In contrast, inherited aetiologies often affect children or young adults. Many have the potential to be partially or completely reversed. Furthermore, novel therapeutics appear promising in the treatment of MONs.
Mechanism of ActionWhether from an inherited mutation or an acquired exposure, overwhelming evidence suggests a shared mechanism of mitochondrial dysfunction. The impairment in ATP production via alternations in oxidative phosphorylation and the accumulation of reactive oxygen species (ROS) within the mitochondria trigger a decrease in the electrical potential across the mitochondrial membrane. This causes cytochrome C to leak out of the mitochondria into the cytoplasm and bind to the apoptosis activating factor-1 (APAF-1) resulting in apoptosis of the RGC.1–4 In Leber hereditary optic neuropathy (LHON), mutations within the mitochondrial DNA lead to dysfunction in the mitochondrial complex I and cause accumulation of ROS. Alcohol, tobacco, and other MON-causing toxins can increase exogenous ROS and thereby cause the same pathway to be activated.1 Whether energy depletion or over-production in ROS precipitates RCG loss remains to be understood and explains the variation in therapeutic targets used.
Susceptibility of prelaminar RGC in MONMON selectively damages the RGCs within the prelaminar area of the papillomacular bundle (PMB). The unique anatomical features of the pre-laminar area make the PMB vulnerable to mitochondrial dysfunction in different ways.5 First, the pre-laminar area contains a high number of mitochondria compared to the post-laminar area, demonstrating that the PMB has a high energy need.6 Secondly, PMB fibers contain a large number of unmyelinated RGC axons compared to the post-laminar area which contains predominantly myelinated axons.2,5 This means that prelaminar axons do not conduct electrical potential in an efficient way compared post-laminar axons which uses saltatory conduction. Thirdly, the PMB fibers are narrow in caliber, which further contributes to decreased energy conduction.5 The high-energy requirement of the PMB in conjunction with the low energy production and slow axoplasmic transport leads to susceptibility of the PMB when mitochondrial dysfunction occurs in MON.2,5,7,8
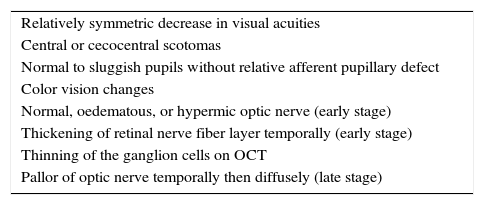
ExaminationClinical PresentationClinical manifestations of MON are summarized in Table 1. Patients with MON typically present with bilateral, painless “fog” at the center of fixation. Visual acuities range approximately from 20/25 to 20/200.9 Only in rare cases such as in methanol toxicity, can vision reach hand motion to no light perception. While visual acuities range widely depending on the severity of the disease, symmetry in visual acuity and ophthalmological findings in both eyes is a hallmark feature of MON.
Clinical manifestations of mitochondrial optic neuropathy.
| Relatively symmetric decrease in visual acuities |
| Central or cecocentral scotomas |
| Normal to sluggish pupils without relative afferent pupillary defect |
| Color vision changes |
| Normal, oedematous, or hypermic optic nerve (early stage) |
| Thickening of retinal nerve fiber layer temporally (early stage) |
| Thinning of the ganglion cells on OCT |
| Pallor of optic nerve temporally then diffusely (late stage) |
The loss of color vision, especially of red color, is a prominent feature of MON. Color vision loss may be more severe than visual acuity loss.2 Hardy Rand and Rittler (HRR) and Fansworth D-15 are most useful because they establish blue-yellow or red-green deficits, whereas Ishihara only assesses red-green defects. Red cap test between two eyes is normal due to the symmetrical nature of genetic or acquired MON. Pupils react equally normal or slightly sluggish to light. Similarly, no relative afferent pupillary defect (RAPD) is observed due to the symmetry of the disease. Reduced contrast sensitivity at high spatial frequencies has been found in sub-clinical cases of MON.10
Ophthalmic fundus evaluation varies depending on the extent of the disease. In early stages of MON, the optic nerves may appear normal, oedematous, or hyperemic. Peripapillary hemorrhages may be present. As the disease progress, PMB loss and temporal optic disc pallor occurs. In patients with longstanding MON, diffuse bilateral optic disc pallor and atrophy is apparent.
Ancillary TestingIn any patient suspected of MON, an Amsler Grid and visual field 30-2 should be performed. In early stages of MON, scotomas may not be present. As the disease progresses, visual field testing and Amsler Grid will reveal bilateral central or cecocental scotomas. Often these defects are more pronounced with a red stimulus compared to a white-on-white stimulus.9
Optical coherence tomography (OCT) is a very useful tool to monitor the subclinical cases and progression of MON. In early stages of MON, OCT may show temporal and inferior-temporal thickening of the retinal nerve fiber layer.11–13 In later stages, thinning especially temporally of the retinal nerve fiber layer and overall ganglion cell layer thinning can be observed1.11,14–17 Visual-evoked potential (VEP) testing may be useful in detecting subclinical MONs. A reduced P100 amplitude with a normal or near-to-normal latency will be observed in MON.18 This helps to differentiate a MON from demyelinating causes of optic neuropathy, such as multiple sclerosis (MS), in which the P100 amplitude is reduced and latency is significantly delayed.18 It should be noted that VEP is a non-specific test as there are multiple aetiologies of reduced amplitudes including amblyopia, inflammatory diseases, and compressive lesions.
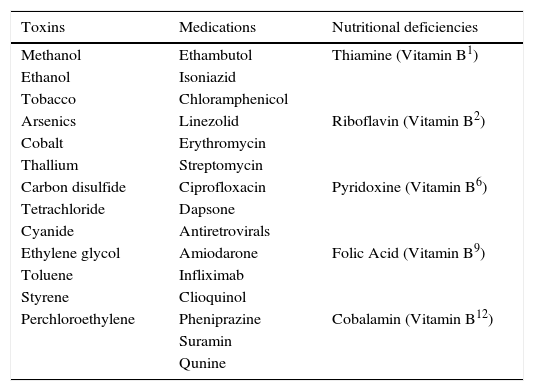
Case HistoryAn extensive case history is essential in a patient suspected of having a genetic or acquired MON. Occupational and social history may suggest possible exposure to toxins. A patient should be questioned about occupational hazards including exposure to fumes (cyanide), organic solvents (ethylene glycol, toluene, styrene, perchloroethylene), heavy metals (lead, mercury, thallium), and carbon dioxide. A social history of alcohol, tobacco, and recreational drug consumption should be documented. Amount, frequency, and duration of intake of substances should be established. It is important to note the type of alcohol consumed since illicit liquor may contain methanol – a life-threatening toxin. In cases of denial of consumption of substances, clinicians should verify if there is a history of substance abuse.
Additional elements of the medical history should be reviewed carefully. As MON can be caused by nutritional deficiencies in vitamin B, establishing a patient's relationship to food is important. Several systemic medications causing MON have been documented (Table 2). Symptoms of toxic peripheral neuropathy, such as numbness or tingling of limbs or gait problems, should be noted. Metabolic diseases, such as diabetes mellitus, thyroid disease, or kidney and liver failure may contribute to exacerbating severity of MON. The presence of a metallic hip replacement should be addressed. Patients with hip-replacements containing cobalt often exhibit symptoms of deafness, poor motor coordination, and numbness of limbs known as arthoprosthetic cobaltism.19–21 A family history of bilateral reduced acuities or blindness may suggest the possibility of a genetic MON.
Causes of acquired mitochondrial optic neuropathies.
| Toxins | Medications | Nutritional deficiencies |
|---|---|---|
| Methanol | Ethambutol | Thiamine (Vitamin B1) |
| Ethanol | Isoniazid | |
| Tobacco | Chloramphenicol | |
| Arsenics | Linezolid | Riboflavin (Vitamin B2) |
| Cobalt | Erythromycin | |
| Thallium | Streptomycin | |
| Carbon disulfide | Ciprofloxacin | Pyridoxine (Vitamin B6) |
| Tetrachloride | Dapsone | |
| Cyanide | Antiretrovirals | |
| Ethylene glycol | Amiodarone | Folic Acid (Vitamin B9) |
| Toluene | Infliximab | |
| Styrene | Clioquinol | |
| Perchloroethylene | Pheniprazine | Cobalamin (Vitamin B12) |
| Suramin | ||
| Qunine |
About 90% of LHON cases are caused by one of three mitochondrial DNA (mDNA) point mutations: m.3460G>A (MTND1), m.11778G>A (MTND4), or m.14484T>C (MTND6). All LHON mutations affect the mitochondrial complex I subunit of the mitochondrial respiratory chain and lead to the accumulation of cytochrome C as described in the common mechanism above.
The minimum prevalence of the three most common LHON mutations is 1 in 31,000 in North East of England.22 Similarly, the prevalence of LHON in the Netherlands has been estimated to be 1 in 39,000.23 Age and sex are the most common risk factors for developing LHON. Approximately 95% of LHON carriers will manifest visual symptoms before the age of 50 years.24 Peak age of onset is usually within the second and third decade.
Not every person identified as having an LHON mutation will develop visual changes. A family history of vision loss in an LHON-carrier may be a predictor of vision loss. But in up to 40% of patients with LHON, no family history is indicated.25 There is a strong sex bias for losing vision in LHON-carriers, known as incomplete penetrance. Approximately 50% of male LHON-carriers and 10% female LHON-carriers lose vision due to LHON.24,25 The causes of incomplete penetrance in LHON-carriers are complex and influenced by numerous genetic, hormonal, and environmental factors.
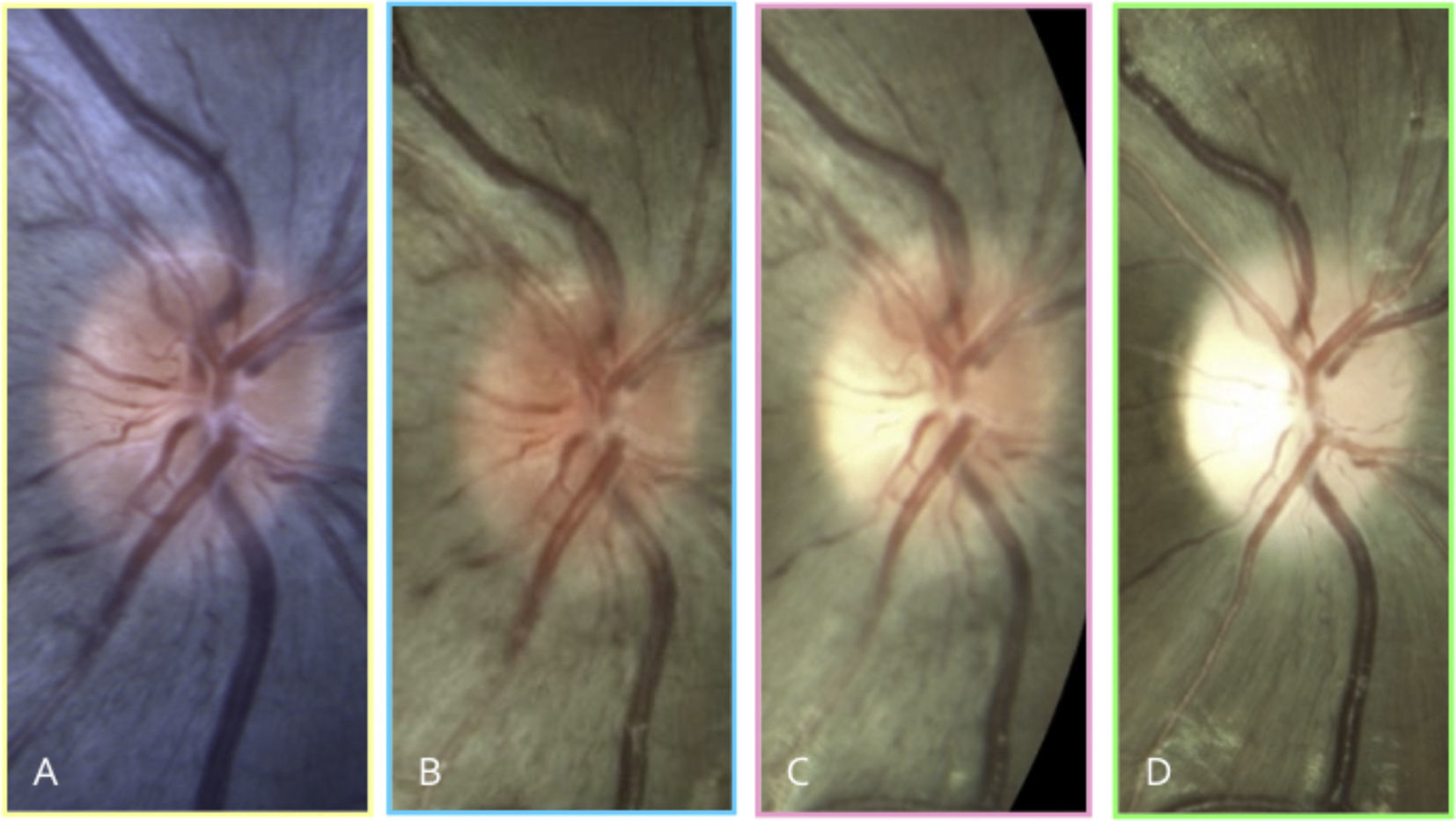
LHON typically manifests with acute unilateral loss of central vision with fellow eye involvement within 2–4 months.8 In about a quarter of LHON patients, bilateral onset of vision loss has been documented. Three clinical features unique to LHON can be observed: (1) vascular tortuosity, (2) peripapillary swelling of the RNFL, and (3) peripapillary telangiectatic vessels with absence of leakage on fluorescein angiography. These clinical changes can be observed even prior to a patient becoming visually symptomatic (Fig. 1).26 However, in about 20% of patients with LHON, optic nerves appear normal during the acute phase.27,28
. At age 12, retinal nerve fiber layer swelling is observed prior to reduction in vision (B). His visual acuity rapidly decreases 1 month later to 20/50 and subtle temporal optic nerve pallor is visible (C). Three months later, his visual acuity is <20/400 and diffuse optic nerve pallor is present (D). Notice that peripapillary telangiectasia was present at age 9 -three years before his visual symptoms started. The patient's left eye progressed similarly to the right eye.")
A 9-year old Asian male with LHON m.11778 presents with best-corrected visual acuity of 20/20 in the right eye and no symptoms (A). At age 12, retinal nerve fiber layer swelling is observed prior to reduction in vision (B). His visual acuity rapidly decreases 1 month later to 20/50 and subtle temporal optic nerve pallor is visible (C). Three months later, his visual acuity is <20/400 and diffuse optic nerve pallor is present (D). Notice that peripapillary telangiectasia was present at age 9 -three years before his visual symptoms started. The patient's left eye progressed similarly to the right eye.
BC 20/20Vision rapidly deteriorates in LHON and prognosis is poor. Within the next six months after onset, visual acuities can reach 20/200 or worse. Chronic signs of LHON include diffuse optic nerve pallor and loss of RNFL. A patient presenting for the first time at this stage in the disease and without family history will be difficult to distinguish from other optic nerve aetiologies.
Environmental causes may contribute to the incomplete penetrance seen in LHON. Some large-scale studies have suggested that smoking and alcohol is associated with the onset of vision loss in LHON-carriers.29,30 Controlling for gender, age, and mutation bias, a European multicentre study involving 196 affected and 206 unaffected LHON-carriers from 125 pedigrees found strong evidence that smoking tobacco was significantly associated with vision loss in LHON-carriers.31 Furthermore, smoking tends to follow a dose-response relationship with heavy smokers having a higher risk of developing vision loss in LHON-carriers. This study also found that consuming alcohol showed a statistically non-significant trend in developing visual changes in LHON-carriers.
Although the overall visual prognosis is poor, patients may spontaneously recover some vision depending upon their genetic mutation. The best chance of spontaneous visual recovery has been observed in patients with m.14484T>C, followed by patients with m.3460G.>A, and the least chance with m.11778G>A. Visual recovery includes improvement in visual acuities, development of small islands of vision surrounding a central scotoma, and recovering color vision.32–34 Furthermore, spontaneous visual recovery is more likely with onset of LHON before 20 years of age, slow progression of visual loss, and large surface area of optic nerves.35
Although LHON primarily affects RGCs, patients with LHON+ phenotype are prone to other medical conditions. There is a higher incidence of cardiac conduction defects, ataxia, peripheral neuropathy, myopathy, and dystonia among LHON-carriers.36 Additionally, there is a condition known as Harding's syndrome in which LHON coexists with a demyelinating syndrome clinically and radiologically similar to MS.37–40
Dominant Optic Atrophy (DOA)DOA, also known as Kjer's optic neuropathy, is a Mendelian inherited optic neuropathy causing mitochondrial dysfunction. The prevalence of DOA within the Caucasian population is estimated to be 1:50,000 with a stronger genetic prevalence in the Netherlands accounting for 1:12,000.41 It has an insidious onset with mild visual loss starting during the first two decades of life. Unlike LHON, patients are asymptomatic in the early stages. Often patients are not identified until testing due to family history or diagnosis of bilateral optic atrophy. Patients often present with visual acuities of 20/30 or better. Although visual prognosis with DOA is highly variable, visual outcome is better compared to LHON with average visual acuities of 20/60 to 20/200.24
Approximately 50–60% of people with DOA have a mutation in the OPA-1(3q28-q29).42,43OPA-1 is a critical pro-fusion protein within the inner mitochondrial membrane. OPA-1 mutations cause mitochondrial defragmentation and lead to respiratory chain dysfunction and dysfunction within the ATPase of the mitochondria.44,45
About 20% of patients with OPA-1 mutations will develop additional co-morbidities of deafness, chronic progressive external ophthalmoplegia, ataxia, myopathy, peripheral neuropathy, and deafness. Deafness appears to be the most common systemic complication often presenting during puberty or young adulthood.46
Other Mitochondrial SyndromesSeveral mitochondrial syndromes with prominent optic neuropathy exist including Charcot-Marie Tooth disease, hereditary spastic paraplegia, Friedrich ataxia, and Costeff syndrome. Although each syndrome has its own characteristics, common signs of these mitochondrial syndromes are distal limb weakness, decreased tendon reflexes, ataxia, myopathy, and deafness.27,33 In patients with suspected mitochondrial syndromes, medical referrals should be initiated immediately due to widespread systemic effects.
Acquired Aetiologies of MONsSubstances and Medication-Induced MONsHeavy tobacco consumption, as well as heavy alcohol consumption, can lead to MON. Due to the co-morbidity of drinking and smoking, the condition was historically named tobacco-alcohol amblyopia – incorrectly named since the condition has no amblyogenic factors.
Methanol, also known as wood alcohol, is considered a very potent toxin capable of rapid onset of MON.47,48 The main cause of poisoning is through adulterated liquor in which methanol is mixed with ethyl alcohol to produce a potent alcoholic drink. Methanol is found in many sources such as windshield wiper antifreeze, canned heating sources, copy machine fluids, deicing fluid, fuel additives, paint remover, and varnish. Toxicity due to methanol may result in symptoms of nausea, vomiting, dizziness, headaches, breathing difficulties, and changes in consciousness.49 Metabolic acidosis may lead to coma and death. Thus, immediate hospitalization is recommended for patients with methanol poisoning. Visual prognosis is also dependent on prompt treatment.
Medication-induced MON may be caused by ethambutol,50–54 linezolid,55–59 chloramphenicol,60,61 erythromycin.24 Less likely to cause MON are ciprofloxacin, streptomycin, isoniazid, antiretroviral drugs, amiodarone, infliximab, clioquinol, dapsone, quinine, pheniprazine, and suramin. Severity of neuropathy depends on dosage and duration.
Nutrition-Deficient MONsNutrition-deficient MON may be caused by deficiencies in thiamine (vitamin B1), riboflavin (vitamin B2), pyridoxine (vitamin B6), folic acid (vitamin B9), or cobalamin (vitamin B12).62–64 In developing countries, the primary cause is undernourishment. In developed countries, vegan diets or weight-loss diets may result in vitamin B deficiencies causing MON.63 In other cases, anorexia nervosa, alcoholism, gastrointestinal (GI) problems or GI surgeries may result in nutritional malabsorption and must be confirmed via blood testing.65 Patients may present with pernicious cell anemia due to vitamin B12 deficiency or Beriberi due to Vitamin B1 deficiency.
Mixed Aetiologies of MONMixed aetiologies of MON add to the complexity of identifying causal agents. Multiple toxins may be responsible for causing an MON as seen in tobacco-alcohol amblyopia. In other cases, toxic exposure may interact with nutritional deficits to cause MON. For instance, the Cuban Epidemic Optic Neuropathy (CEON) outbreak in 1991–1993 affecting 500,000 people resulted from malnutrition due to the country's economic problems and a high prevalence of tobacco, particularly cigar smoking.66–68 Furthermore, genetic MONs may be exacerbated or even triggered by toxic or nutritional causes of MON.
Differential DiagnosisA comprehensive medical and social case history, as outlined previously, may indicate potential causes of MON. However, MON is a diagnosis of exclusion and often multi-factorial. Thus, any case of suspected MON, an MRI of the brain and orbit is essential to rule out other aetiologies.
Compressive and demyelinating lesions may cause oedematous or atrophic optic nerves. Symptoms of a compressive lesion may include headache and changes in mood, personality, or memory. An MRI of the brain and orbits will confirm diagnosis.
Optic neuritis may be differentiated from MON due to the pain upon eye movement. Visual recovery within weeks further confirms the diagnosis of optic neuritis, whereas visual acuity loss and central scotomas will progressively worsen in MON. A brain MRI should be ordered due to the strong association between optic neuritis and MS. As mentioned previously, MON can co-exist with in a MS-like syndrome known as Harding's syndrome. Diffuse white matter lesions on MRI suggest MS. VEPs also help differentiate MON from demyelinating causes of optic neuropathy in which the response is delayed and P100 amplitude is reduced.18
Ischemic optic neuropathy presents with oedema of the optic nerves in the acute stage and diffuse optic atrophy in the chronic stage. In cases of bilateral optic nerve oedema the possibility of increased intracranial pressure (ICP) – a life-threatening condition – must be considered. Presence of spontaneous venous pulsation within the optic nerves may indicate normal ICP. However, the absence of venous pulsation occurs in about 30% of patients and if not previously documented is not conclusive. Symptoms of increased ICP may include headache, dizziness, nausea, and vomiting. In patients over 55 years, an artertic ischemic optic neuropathy (AION) should be ruled out by ordering C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and potentially a temporal artery biopsy. Patients will normally report symptoms of jaw claudication, temporal headaches, and sensitivity to touch on temporal side of head.
Bilateral traumatic optic neuropathy may be suspected with a history of reported head or eye trauma. In these cases, identifying trauma to other eye structures can be important in making diagnosis.
Normal Tension Glaucoma (NTG) and MONs share similar clinical characteristics of insidious visual acuity loss, paracentral defects, and thinning of neuroretinal rim. Particularly, overlapping commonalities between NTG and DOA have lead some researchers to postulate whether or not NTG is a hereditary optic neuropathy. Multiple studies have found an association between OPA1 polymorphisms and normal tension glaucoma (NTG).69–71 However, NTG manifests itself in older population with an average of approximately 64 years.72 Arcuate visual field defects, excavation of the inferior rim, and inferior optic nerve thinning also differentiate NTG from MONs.73
Differentiating an aetiology of MON requires different blood tests. Laboratory blood tests for vitamin B1, B2, B6, B9, and B12 may establish nutritional causes for MON. Patients with substance abuse should also have laboratory tests for nutritional deficiencies, as these two MON aetiologies often co-exist. If heavy metal toxicity is suspected, a 24-h urine test to measure exposure to heavy metals can be conducted. A family history of bilateral vision loss may suggest a genetic aetiology of MON and may warrant further genetic testing. It should also be remembered that toxic or nutritional causes of MON may exacerbate a pre-existing genetic MON.
Optometric Management of MONBaseline Testing and MonitoringBaseline vision testing includes visual acuities, color vision, Amsler grid, visual fields, and ophthalmic evaluation of optic discs and retinal nerve fiber layer, and documentation with photography or OCT. Genetic testing to rule out LHON and DOA should also be initiated.
Follow-up examinations are recommended every 4–6 weeks in patients with reduced vision, a family history of LHON or DOA, or patients with a strong suspicion of MON. Even if the patient is asymptomatic, close monitoring every 4–6 weeks is essential. Patients should be strongly urged to return to clinic immediately once they notice a change in vision. Furthermore, medical referrals may be necessary if substance abuse, medication toxicity, or nutritional deficits are suspected. For suspected LHON+ or DOA+ phenotypes, immediate medical referrals to address cardiovascular, neurological, motor or ear problems should be initiated.
Cessation of Agent inducing MONDiscontinuation of causal agent (e.g. medication, tobacco, alcohol, methanol) is recommended. In acquired MON, discontinuation of toxicity-causing factor can lead to recovery of visual acuity and central vision. Even with significant loss of the PMB, visual acuities can often remain on average 20/50 after recovery and full visual recovery from substance and medication-induced MONs has been documented in the literature.
Genetic CounselingGenetic testing is indicated for family members of a patient with an inherited MON. Individuals identified with the mutation should undergo baseline testing as mentioned. Genetic counseling may also be beneficial for individuals with an inherited MON who are planning on starting a family.
Clinical TrialsIntroducing a patient to current clinical trials is an option. Particularly, a patient with an inherited MON may benefit from this treatment option. Generally speaking, early therapeutic intervention is recommended to lessen the chance of irreversible damage.
Facilitating Access to Rehabilitation Resources and ServicesUnfortunately patients with MONs, especially those with LHON, have a poor visual prognosis. Often LHON patients are children or young adults who are otherwise systemically healthy. Rapid vision loss is devastating and needs to be properly addressed by the managing optometrist. Facilitating access to rehabilitative services, such as low vision and occupational therapy, can be valuable to a patient adapting to vision loss. Family involvement and support during this period is recommended.
Therapeutic InterventionsCoenzyme Q10 (CoQ10)Various pharmacological targets are being explored to mitigate the mitochondrial dysfunction that occurs in MON. Quinone analogs, such as CoQ10, help restore the electron chain transport and promote ATP synthesis within the mitochondria. They do this by bypassing mitochondrial complex I inhibition and shuttling electrons from the cytosol directly to complex III, thereby re-establishing ATP synthesis and ultimately reducing cytochrome C.8,74 Historically, CoQ10 has been used in mitochondrial disease due to its cellular mechanism of action.
Short-Chain Quinones: IdebenoneRecent studies have shifted attention toward idebenone and EPI-743, two shorter chain quinones, because they may be more effective than CoQ10. Due to its lipophobic properties, these two quinone analogs when orally administered are able to be delivered more readily into the mitochondria and have been reported to be more potent.
Over the last decade, studies have focused on idebenone therapy for the treatment of LHON. In 1992, the first case report was published reporting that 10-year-old boy with the LHON showed visual recovery with a low daily dosage of 90mg idebenone.75 The Rescue of Hereditary Optic Disease Outpatient Study (RHODOS), a multi-center, double-blind, randomized, placebo-controlled study was conducted to investigate if daily dosages of 900mg idebenone for 6 months help slow progression in 85 patients with LHON mutations m.3460G4A and m.11778G4A.76 Although the RHODOS study failed to show a statistical difference in visual acuity between the two groups, idebenone treatment showed a trend in maintaining visual acuity over 6 months. Furthermore, patients with unequal visual acuities were more likely to benefit from idebenone treatment, suggesting early treatment with idebenone may be important in preventing fellow eye involvement. In the follow-up study, visual acuity remained stable even 30 months after discontinuation of the of the 6-month long idebenone treatment.77
The same year, a retrospective study by Carelli et al. additionally demonstrated a trend in visual recovery in patients using various dosages of idebenone within the first year of disease onset compared to untreated LHON patients.78 Interestingly, LHON-patients with the mutation m.11778/ND4 showed statistically significant visual recovery when treated with idebenone whereas patients with m.3460/ND1 did not respond as well.
Differences in the two large studies of idebenone in 2011 may be of clinical value. Carelli et al. only included LHON-patients who started idebenone treatment within 1 year after onset of the disease to control for the spontaneous recovery, which occurs mostly in 2–5 years after LHON onset. The RHODOS study, on the other hand, included LHON-patients with onset of disease up to 5 years ago. Treatment with idebenone in RHODOS patients may have not been as effective due to irreversible damage to RGCs with prolonged period of the disease. Thus, the importance of immediate treatment with idebenone is stressed once diagnosis of LHON has been made.
While treatment onset, duration, and dosing differed between these two studies, both support the therapeutic effect of idebenone for LHON. As a result, the European Medicines Agency (EMA) approved idebenone under the tradename Raxone® in September 2015 for the treatment of LHON.
The promising findings of idebenone in LHON treatment have spurned interest in exploring the efficacy of idebenone in DOA. In a small pilot study, seven DOA patients with OPA-1 mutation received variable doses of idebenone (270–675mg/day) for at least one year.79 Five out of seven idebenone-treated patients showed an increase in visual acuities and four out of seven idebenone-treated patients demonstrated an improvement in color vision. A large-scale, randomized, placebo-controlled study is needed to fully evaluate visual recovery of DOA-patients using idebenone.
Short-chain Quinones: EPI-743Another short-chain quinone, EPI-743, has shown promise in the treatment of LHON. In an open-labeled clinical trial, four of five LHON-patients treated with EPI-743 within 90 days after disease onset for a period of at least 1 year, showed variable improvements in visual acuity or visual field.80 Studies to further explore the effects of EPI-743 in patients with DOA are needed.
Mitochondria-Targeted QuinonesTo increase delivery into the mitochondria, short-chain quinones have been coupled to the lipophiliccation, triphenylphosphonium (TPP). Mitoquinone mesylate (MitoQ) and 10-(6′-plastoquinonyl) decyltriphenyl phosphonium (SkQ1) have been suggested as potential therapeutics. However, more evidence is needed to substantiate these claims.81
Gene TherapyGene therapy has shown potential in the treatment of LHON. In vitro LHON studies have focused on the allotopic expression of mitochondrial-encoded genes delivered via an adeno-associated virus (AVV) vector into the mitochondria.82,83In vivo studies have also demonstrated that intravitreal injections of the AVV-mediated ND4 wildtype gene improves visual function in rodents.84,85 However, it remains controversial due to the questionable feasibility of integration of ND4 wildtypes subunit into the mitochondrial membrane.86 Recently, two other genetic engineering strategies have shown promising results. Direct delivery of circular mDNA into the mitochondria demonstrated restoration of cellular respiration and ATP production in vitro.87 Another genetic approach involving an AAV-vector containing mDNA encoded with wildtype ND4 subunit tag showed successful expression within the inner retinal layers and prevention of visual loss and optic atrophy in the LHON-mutant R340H ND4 mouse model.88
The groundwork has been laid for clinical gene therapy studies and promising results are emerging. In an open labeled prospective study, five patients with the LHON carrying the m.11778G>A mutation were evaluated after an intraocular injection of an AVV-mediated ND4 construct was given.89 While the visual acuity of three of the participants remained unchanged after three months, two of the five patients experienced moderate improvements in visual acuity. No serious adverse effects were reported. In a related 9-month follow-up study, visual acuity of six of the nine patients’ treated eye improved by at least 0.3log MAR.90
Other gene therapy clinical trials are currently in progress. GenSight Biologics is currently sponsoring an open label trial (NCT02064569) to investigate the safety and tolerability of intraocular injections of AAV2-mediated ND4 in 21 LHON patients with the m.11778G>A mutation. Additionally, the company will evaluate the visual outcome of gene therapy in LHON patients with same mutation when vision loss is present (a) six months or less (NCT02652767) or (b) six to twelve months (NCT02652780). Another clinical trial conducted by the University of Miami will investigate the safety in 27 patients with LHON using different doses of the AAV-ND4 gene therapy (NCT02161380) and is scheduled to be completed in March 2019.
Gene therapy for treatment of DOA is in its infancy. It is well known that DOA is linked to OPA-1 mutations and rodent models carrying vision-deficient OPA-1 mutations have been generated.91–93 One approach is trying to develop intravitreal injections of an AAV vector to deliver wildtype OPA1 to target RGCs in a DOA mouse model.
Antioxidants: MTP-131MTP-131, also known as Bendavia, is a Szeto–Schiller (SS) peptide that selectively targets the inner mitochondrial membrane. It acts as a potent antioxidant and enhances electron transport chain function. Since it has shown potential in the treatment of MON clinical studies are currently underway. A phase 2 clinical trial aims to evaluate if a 1% ophthalmic solution of MTP-131 over 16 weeks is a safe and effective treatment for 12 subjects with LHON (NCT02693119).
Other Therapeutic TargetsOther therapeutic targets, out of the scope of this paper, have shown potential in the treatment of MON. They include aestrogen, methylene blue, cyclosporine A, brimonidine, near-infrared therapy, and stem cell therapy.36,83,94
ConclusionMON is an umbrella term used to describe inherited and acquired optic neuropathies with the same clinical manifestations. All MONs exhibit selective loss of the RGCs due to mitochondrial dysfunction. Many acquired MONs are reversible and various novel therapeutics are on the horizon as treatment options.
FundingNo funding sources.
Conflict of interestNo conflict of interest is present.





